McCune-Albrighti sündroom sai nime kahe silmapaistva arsti järgi, kes kirjeldasid seda rohkem kui pool sajandit tagasi. Nad rääkisid ühiskonnale lastest, kellest enamik olid tüdrukud. Paljudel neist oli lühike kasv, ümar nägu, lühike kael, lühenenud IV ja V pöialuud või kämblaluud, lihasspasmid, luustiku muutused, hilinenud hammaste tulek ja emaili hüpoplaasia. Samuti oli vaimne alaareng ja endokriinsed haigused mida väljendab varajane puberteet koos menstruaalverejooksuga, rindade areng, häbeme- ja rinnakarva kasv, laste kasvutempo kiirenemine ja nahamuutused.
Kaasaegses meditsiinis kasutatakse terminit "Albrighti sündroom" patsientide puhul, kellel on kõik või ainult mõned endokriinsüsteemi ja naha kõrvalekalded. On juhtumeid, kui diagnoos tehti varases lapsepõlves. Kuid tüüpilistel juhtudel antakse seda 5-10-aastastele lastele, võttes aluseks sellele haigusele iseloomulikud tunnused. Üldiselt on see haruldane ja pärilik. Selle haiguse etioloogia ja patogenees on endiselt teadmata.
Sündroomi põhjused
Sündroomi etioloogia pole teada. Eeldatakse neurogeenseid mõjusid ja endokriinseid mutatsioone, mis on põhjustatud hüpotalamuse piirkonna kaasasündinud kõrvalekalletest embrüonaalse arengu ja gonadotroopse hormooni varase tootmise ajal. Teadlased usuvad, et McCune-Albrighti sündroomi arengu põhjustab neeru- ja luurakkude vähene reaktsioon hormoonide toimele. kõrvalkilpnäärmed.
Kaltsiumi ja fosfori taset vereringesüsteemis reguleerivad kõrvalkilpnäärmed, kuid selle patoloogilise seisundi korral põhjustab nende puutumatu (ükskõikne) reaktsioon nende elementide kõikumisele hüperparatüreoidismi (paratüreoidhormooni liigne tootmine), luu struktuuri muutusi. . Neerudes on häiritud cAMP, tsüklilise adenosiinmonofosfaadi, üksikute hormoonide, sealhulgas paratüreoidhormooni signaalide levimise abilüli, sünteesi normaalne kulg.
McCune-Albrighti sündroomi korral põhjustab ebaõnnestumise geenimutatsioon. Rikkumine võib esineda kõikjal metaboolses ahelas: mõnel patsiendil on retseptor kahjustatud, paljudel patsientidel on rakumembraani valgud muutunud, teistel on cAMP-i tootmine ebapiisav. Neid juhtumeid ühendav geneetiline seisund ei ole päritav: mutatsioon tekib pärast viljastamist.
Sümptomid
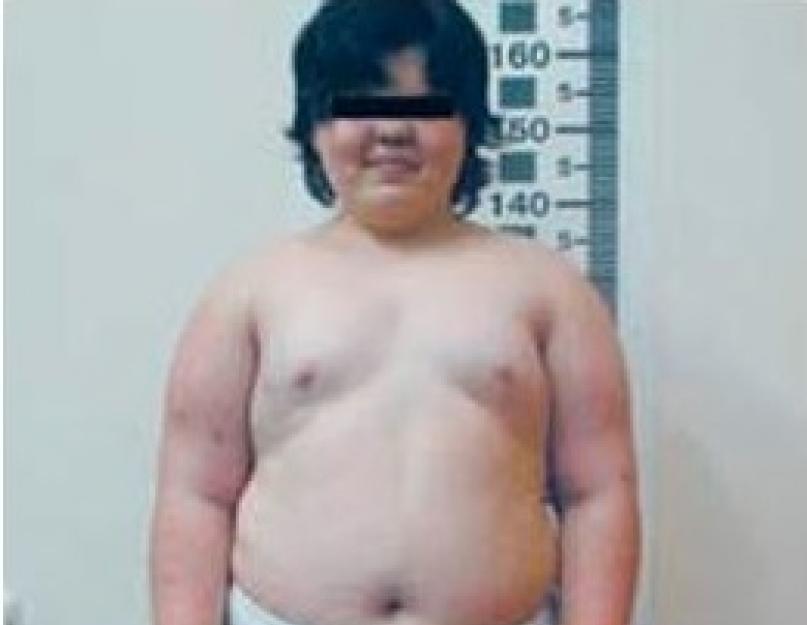
Tüüpilised nähtavad märgid on järgmised:
- madal kasv;
- ülekaal;
- kõrge glükoosisisaldus;
- kuu nägu;
- puberteedi häired.
Samuti võivad ilmneda järgmised sümptomid:
- akromegaalia;
- autoimmuunhaigused;
- günekomastia;
- Itsenko-Cushingi tõbi jne.
Patsiendid kurdavad sageli verd uriinis, pidevat oksendamist, muutusi hammastes, krampe. Uurimisel on näha subkutaanset luustumist, võivad ilmneda kaltsifikatsioonid.
Sageli on patsientidel lühenenud sõrmed, toruluude epifüüsid on deformeerunud, võib esineda läätsekujulise katarakti tunnuseid.
Peaaegu alati on sellised patsiendid vaimses arengus maha jäänud.
Haiguse diagnoosimine
Haiguse diagnoosimisel viiakse läbi järgmised uuringud:
- Kirjeldatakse kliinilisi ilminguid;
- Albrighti sündroomiga patsient saadetakse uuringule geneetiku juurde;
- röntgenuuring;
- hormoonide testid (sugu, kilpnääre, gonadotropiin ja teised vastavalt näidustustele);
- Vaagna ultraheliuuring;
- Endokrinoloogi, günekoloogi (tüdrukutele), uroloogi (poistele) läbivaatus;
Vajadusel elundiuuring endokriinsüsteem.
Albrighti sündroomiga inimesed on registreeritud mitmete spetsialistide juures, kes vajavad jälgimist (günekoloog - endokrinoloog, spetsialist radiodiagnoos, ortopeediline traumatoloog). Lisaks arstide külastamisele peavad patsiendid perioodiliselt võtma hormoonide (troopiliste, gonadotroopsete) teste.
Patoloogia ravi
Albrighti sündroom on ravimatu haigus. Et säilitada patsientide elu optimaalsel tasemel ja kõrvaldada peamised kliinilised tunnused haigusi ravitakse sümptomaatiliselt. Kui üldisi ravimeetmeid ei võeta õigeaegselt, vaimne alaareng ainult süveneb.
Moodustamisele aitab kaasa individuaalselt koostatud dieediteraapia vähendatud fosforisisaldusega dieedis normaalne tase kaltsium veres.
Sündroomi ilmingute kompleksne ravi hõlmab:
- kaltsiumipreparaadid - Calcium D3 Nycomed, Calcemin, Calcemin Advance;
- ravimid, mis seovad fosforit ja aeglustavad selle imendumist soolestikus - kaltsiumatsetaat, Nefrosorb, Renacet;
- D-vitamiini sisaldavad tooted - "Calcitonin", "Miacalcic", "Sibacalcin";
- antiandrogeenid - "Diana-35", "Zhanin", "Yarina";
- progesterooni preparaadid - "Utrozhestan", "Dufaston";
- ravimid, mis pärsivad hormoonide sünteesi kilpnääre- "Tiamasool", "Merkasoliil";
- vahendid, mis vähendavad kortisooli sünteesi - Bromkriptin, Trilostan, Mitotan;
- ravimid, mis pärsivad kasvuhormooni tootmist - Somatulin, Oktreotiid, Sandostatin;
- aromataasi inhibiitorid - Aromasin, Egistrazol, Estrolet;
- krambivastased ained - "kaltsiumglükonaat", "kaltsiumkloriid".
Liigse fosfori edukaks eemaldamiseks organismist on igapäevane hemodialüüs end hästi tõestanud.
Akromegaalia korral tehakse kirurgilist ravi. See on näidustatud ka progresseeruva nägemiskahjustuse, tugeva valu ja tõsiste vigastustega patsientidele. Kaasasündinud või omandatud luudefektide korrigeerimiseks tehakse luusiirdamine. Kasvaja või tsüst kraabitakse luust välja ilma terveid kudesid mõjutamata.
Ärahoidmine
Ennetamine on järgmine: sellise patoloogiaga inimesed peaksid regulaarselt tegema füüsilised harjutused lihaste tugevdamiseks; kui naine soovib rasestuda, siis on vaja konsulteerida günekoloogi ja geneetikuga. Seda patoloogiat diagnoositakse ühel inimesel miljoni planeedi elaniku kohta. Spetsiifilist ravi kahjuks veel ei ole. On ainult sümptomaatiline ravi. Kuid kui patsient on arsti järelevalve all, on soodsa tulemuse võimalus.
McCune-Albrighti sündroom on heterogeenne haigus, mida kirjeldasid sõltumatult Ameerika lastearst D. J. McCune ja endokrinoloog F. Albright aastatel 1936–1937. Haigus on haruldane, kaasasündinud, kuid mitte pärilik, millega kaasneb ainevahetushäired, enneaegse seksuaalse arengu tunnused ja luustiku deformatsioonid. Esimesi kliinilisi ilminguid täheldatakse varases lapsepõlves või noorukieas arenguhäirete, polüostootilise kiulise luudüsplaasia, naha pigmentatsiooni nagu "piimakohv" ja endokriinsete anomaaliate, sealhulgas Cushingi sündroom , .
Albright McCune'i sündroomi diagnoositakse tüdrukutel ligikaudu 10 korda sagedamini kui poistel. Wikipedia märgib, et esinemissagedus inimpopulatsioonis on 1 juhtu 100 tuhande kohta ehk miljoni inimese kohta.
Patogenees
Geeni postsügootse mutatsiooni tulemusena GNAS1 osaleb rakuvalgu signaaliülekandes GS-α pakkudes luteiniseerivate ja folliikuleid stimuleerivate hormoonide retseptorite sidumist molekulidega adenülaattsüklaas munasarjade tsütoloogilistes struktuurides. Mutantse valgu pidev mõju aktiveerib adenülaattsüklaasi ja suurendab rakusisest laager ning stimuleerib ka steroidsete naissuguhormoonide suurenenud sekretsiooni folliikulite östrogeeni tootvate tsüstide poolt, põhjustades enneaegset seksuaalne areng isegi puudumisel. Sama mustrit täheldatakse ja "autonoomset tootmist" täheldatakse kilpnäärme hormoonide puhul ja. Patoloogia progresseerumist põhjustab juba mutantseid valke sisaldavate rakkude moodustumine.
Lisaks põhjustab cAMP taseme tõus tootmise suurenemist ja läätsede laikude moodustumist, fokaalset hüperpigmentatsiooni normaalväärtused ja melanotropiin .
Fibrotsüstiline düsplaasia östrogeenide toimel suurenenud lupjumise tagajärjel toimub see normaalse asendamisega luu struktuurid ja moodustuvad kiuliste masside poolt kaltsifikatsioonid, samas on häiritud luude resorptsiooni ja regeneratsiooni protsesside tasakaal, mesenhümaalsete stroomarakkude proliferatsioon. Täheldatakse hävitamise ja pseudotumorite fookuste teket.
Klassifikatsioon
Albrighti tõbi on erinev, sõltuvalt kursi tõsidusest, eristavad nad:
- varjatud haigus - ilma luu- või endokriinsete patoloogiate tunnusteta, samuti ebatavalise pigmentatsiooni sümptomiteta;
- sündroomi mittetäielikud vormid , näiteks luukoega seotud patogeneesi protsessis on võimalikud endokriinsed muutused ja pseudohüpoparatüreoidism kuid nahailmingud puuduvad;
- pseudohüpoparatüreoidism või pärilik Albrighti osteodüstroofia - haigus, mis mõjutab ka luukoe, kuid mehhanism ja etioloogia on erinevad - patoloogia on põhjustatud resistentsuse rikkumisest paratüreoidhormoon , kaltsiumi ja fosfori metabolism; pseudohüpoparatüreoidism avaldub ka lapsepõlves ja väljendub lühikese kasvu, ümara näo, vaimse alaarengu kujul, hüpogonadism , jne.

Põhjused
Albright McCune'i sündroom tekib embrüogeneesi varases staadiumis geneetiliste häirete tagajärjel. Siiani ei ole pärandi tüüp kindlaks tehtud, kõik kirjeldatud juhtumid olid juhuslikud.
Sümptomid
Haiguse kliiniline pilt avaldub terve sümpatokompleksi kujul, mis koosneb:
- enneaegne gonadotropiinist sõltumatu seksuaalne areng, mis areneb hiljem ja aeglasemalt kui teiste varase puberteedi vormide puhul;
- naha laiguline pigmentatsioon - lentigo , helepruuni värvi selgete piiridega laigud esinevad tavaliselt sünnist saati rinnal, seljal, alaseljal, puusadel ja luude deformatsiooni kohtades;
- endokriinsed häired, mis põhjustavad seletamatut kehakaalu langust, silma sümptomid hüpertüreoidism ;
- polüostootiline fibroosne osteodüsplaasia , ja, kaasates protsessi sagedamini pikki luid, ilmnevad rikkumised moonutatud näojoonte, pikliku kehaehituse, väändunud ja asümmeetrilise, lühenenud randmete kujul, mis raskendab kõndimise ajal liikumist, põhjustades lonkamist, äge valu ja suurendab patoloogiliste luumurdude tõenäosust.

Esimesed enneaegse puberteedi tunnused algavad emakaverejooksuga, millel ei ole tsüklilisust ja mis on põhjustatud östrogeenide ja gonadotroopsete hormoonide kontsentratsiooni lühiajalisest tõusust vereringes. Sellised kõikumised provotseerivad tsüstide moodustumist munasarjades. 5-10 aastastel lastel rinnanäärme varajane kasv ja günekomastia . Selle õrn etioloogia ei põhjusta sekundaarsete seksuaalomaduste teket - McCune-Albright-Braytsevi sündroom ei põhjusta varajast seksuaalset karvakasvu ega figuuri diferentseerumist. Tõeline puberteet on võimalik pärast puberteedi lõppu.
Analüüsid ja diagnostika
McCune'i sündroomi kahtluse korral viiakse Albright läbi:
- Röntgenuuringud, mis näitavad pseudotsüstid , skeleti mis tahes osade segmentaalne kahjustus - sagedamini alajäsemed, harvematel juhtudel - ülemine õlavöö ja kolju;
- ultraheli kõhuõõnde võimaldab tuvastada;
- tuvastamiseks laboratoorsed testid edasijõudnute tase aluseline fosfataas vereringes, gonadotropiinide vähenemine uriinianalüüsides ja suurenenud eritumine luteiniseeriv hormoon , 17-hüdroksü ja 17-hüdroksü kortikosteroidid .
Ravi
Ravi on sümptomaatiline, sellel on oma raskused sõltuvalt individuaalsetest omadustest ja kliiniline pilt. Analoogid gonadoliberiin ei oma terapeutilist toimet, kuna östrogeeni sekretsiooni protsessi ei provotseeri gonadotropiinide liig. See võib olla tõhus ka meestel sugunäärmete steroidogeneesi peatamiseks. Ravi ajal medroksüprogesteroonatsetaat võimalik areng hüpokaltseemia Seetõttu, kui varase puberteedi arenguga kaasnevad luukoe rasked mitmed kahjustused, tuleb ravi ette näha ettevaatusega.
Rõhumise eest hüperestrogenisatsioon ja võib välja kirjutada skeleti pikendavaid ravimeid. Luu düsplaasia vastu võitlemiseks peate sisestama:
- bisfosfonaadid ;
- osteoklastide antagonistid.
Patsiendid vajavad regulaarselt, iga 3 kuu tagant. kilpnäärme ja teiste näärmete probleemide lahendamiseks külastage endokrinoloogi.
Arstid
Ravimid
- – hoolimata sellest, et see on rohkem tuntud seenevastase ravimina, on see suurtes annustes efektiivne Cushino-laadsete ilmingute korral. Päevane annus, jagatuna 2-3 annuseks, võib ulatuda 1200 mg-ni.
- Medroksüprogesteroon - on androgeense, kasvajavastase ja anaboolse toimega. Raviarsti poolt individuaalselt määratud annustes on kehasse viimiseks erinevaid viise.
- pamidroonhape - ravim on võimeline ära hoidma luude osteolüütilist hävitamist. Võib manustada intravenoosse boolusena, kuid seda ei tohi kombineerida raviga teiste bisfosfonaatidega.
- - bisfosfonaat, mis aitab kaasa normaalse histoloogilise struktuuriga luude moodustumisele, nende taastumisele ja mineraalse tiheduse suurenemisele. Ravimit tuleb võtta suu kaudu sees - 1 tablett nädalas.
Protseduurid ja toimingud
- Munasarjade eemaldamine - Hüperpuberteedi ülemäärase progresseerumise korral võib soovitada steriliseerimist.
- Skeleti ja kolju luude deformatsiooni kirurgiline rekonstrueerimine.
Prognoos
Viimastel aastatel on hakatud üha sagedamini rääkima McCune Albrighti sündroomist ja selle ravi probleemidest ning seda haigust põdevad inimesed hakkasid isiklikes blogides ja videotes end deklareerima ning igapäeva- ja ühiskondliku elu jooni paljastama. Tänu kaasaegsed tehnoloogiad, vaatamata patoloogia ravimatusele on selliste inimeste elukvaliteet oluliselt tõusnud, nüüd võivad nad leida eneseväljendusviisi ja isegi filmides näitleda, näiteks Uruguay näitleja Mauricio Saravia kombel.
Allikate loetelu
- McCune. D. J. Osteitis fibrosa cystica: üheksa-aastase tüdruku juhtum, kellel on samuti enneaegne puberteet, naha hulgipigmentatsioon ja hüpertüreoidism. American Journal of Diseases of Children, Chicago, 1936; 52:743–747.
- Mandel B. R. Kaasaegse geneetika alused. Õpetus. M: Flinta, 2017. - 305 lk.
- Kulakov V.I., Manukhina I.B., Saveljeva G.M. Günekoloogia: nat. käed M. : GEOTAR-Meedia, 2011.- 461 lk.
Sünonüümid: Albrighti sündroom, polüostootiline kiuline düsplaasia.
Definitsioon. Pärilik haigus, mis väljendub naha pigmentatsioonihäirete koos kiulise luudüsplaasia ja endokriinse patoloogiaga.
Ajaloo viide. Esimest korda selle haiguse kohta avaldas A. Weill 1922. aastal, kirjeldades 9-aastast tüdrukut, kellel oli enneaegne puberteet, haprad luud ja naha pigmentatsioon. 1932. aastal avaldati sellele sündroomile iseloomulikud muutused (Vera Gaupp). Lisaks kirjeldas haigust Ameerika lastearst D.J. McCune (Donovan James McCune) 1936. aastal ja Ameerika endokrinoloog F. Albright jt 1937. aastal. Fuller Albright jt teatasid 1937. aastal 21 vaatluse põhjal süsteemsest haigusest, mida nad nimetasid "sündroomiks, mida iseloomustab dissemineerunud fibroosne osteiit". , pigmendiväljad ja endokriinsed häired koos enneaegse puberteediga tüdrukutel.
Etioloogia ja patogenees. Sündroomi põhjustavad mutatsioonid GNAS1 geenis, mis paikneb 20q13.2 lookuses. Geen kodeerib guaniini nukleotiidi siduvat valku (G-valku). Pärandi tüüp pole teada. Sündroom ei ole pärilik. Mutatsioonid on juhuslikud ja esinevad loote arengu ajal.
Sagedus. Sündroom on suhteliselt haruldane (1:100 000 ja 1:1 000 000 elanikkonnast).
Vanus ja sugu. Nahamuutused esinevad sagedamini tüdrukutel ja tavaliselt kohe sündides.
Nahakahjustused. Vahetult pärast sündi või vahetult pärast seda ilmuvad nahale geograafilise kaardi tüüpi ebakorrapäraste piirjoontega helepruunid laigud, sakiliste servadega “pearanniku” kujul, reeglina suured. Tumedad laigud paiknevad sageli laikudena ja on rohkem väljendunud kahjustatud poolel, eriti sageli kuklal, seljal, puusadel, nimmepiirkonnas.
Luu patoloogia. Osteoporoosi mitmekordsed tsüstilised kolded koos osteoskleroosi ja hüperostoossete piirkondadega (eriti kolju luudes). See põhjustab luude patoloogilist haprust, pseudoartroosi, luuaukude deformatsiooni, mis põhjustab neuroloogilisi sümptomeid, valu. Pikkadel luudel on eelsoodumus kõverdumiseks. Kolju ja näo luustiku deformatsioon võib olla märkimisväärne ja sellega kaasneda nägemise ja kuulmise halvenemine. Luu haaratus on tavaliselt asümmeetriline ja võib olla haiguse ainus sümptom.
Endokriinne patoloogia. Tüdrukutel tekivad varase puberteediea tunnused (menstruatsioon ja sekundaarsed seksuaalomadused ilmnevad sageli 7-aastaselt). Poistel on seksuaalne areng normaalne, neil võib esineda hüpergenitalismi või sugunäärmete atroofiat. Patsientidel võib olla ka hüpertüreoidism, hüperparatüreoidism, hüperprolaktineemia, hüpersomatotropism, Itsenko-Cushingi sündroom.
Diagnoos panna põhjal olemasolu värvi laigud "", luu patoloogia, enneaegne seksuaalne areng, hüpertüreoidism, anomaaliad neerupealiste, akromegaalia. Kell laboriuuringud näitavad kilpnäärmehormoonide, kõrvalkilpnäärmete, neerupealiste, samuti kasvuhormooni ja prolaktiini taseme tõusu. Röntgenuuringul avastatakse kiuline düsplaasia, mis mõjutab mitut luud. MRI võib paljastada hüpofüüsi adenoomi. Geneetiline testimine tuvastab GNAS1 geeni.
Eristada grupis pärilikud haigused, mis näitavad ka kohviku-au-lait kohti. Kohviplekid ei ole selle sündroomi usaldusväärne tunnus, kuna neid leidub sagedamini tervetel inimestel ja neurofibromatoosiga patsientidel. Kuid McCune-Albrighti sündroomiga patsientidel on café-au-lait laigud tavaliselt suured, ebakorrapäraste piirjoontega, sakiliste servadega ja paiknevad peamiselt luupatoloogia läheduses.
Kursus ja prognoos. Haigus on kliiniliselt heterogeenne. Klassikalise märkide triaadi juhtumite kõrval on sündroomi ebatüüpilised ja mittetäielikud vormid. Sümptomite raskusaste ja haiguse raskusaste on samuti väga erinevad. Vanusega luukoe patoloogia progresseerub. Ühe luupatoloogia areng ületab sageduselt kogu sümptomite kompleksi 30-40 korda. Pahaloomulised kasvajad harva (vähem kui 1%). Reeglina leitakse osteosarkoome, eriti RT, mõnikord kilpnäärme-, munandi- ja rinnavähi taustal.
Ravi sümptomaatiline. Östrogeeni sünteesi inhibiitorid võivad pärssida enneaegset puberteeti.
- Naaske jaotise pealkirja "
Albrighti sündroom on ebaselge päritoluga pärilik patoloogia. Seda nimetatakse ka pseudohüpoparatüreoidismiks. Seda iseloomustab luukoe kahjustus. Haigus avaldub peamiselt naissoost pooltel elanikkonnast, kuigi esineb ka poistel. Peaaegu kõigil patsientidel esineb kõrvalekaldeid vaimses ja füüsiline areng. Inimese luud hakkavad mõju all lagunema metaboolsed protsessid fosfor ja kaltsium. Algab paratüreoidhormooni vabanemine kõrvalkilpnäärme poolt.
Etioloogia
Sellise patoloogia nagu Forbes-Albrighti sündroomi peamine põhjus on keha kudede pärilik resistentsus paratüreoidhormooni mõjule. Kui inimene ei ole haige, siis toimib hormoon teatud aine kaudu. Kui haigus avaldub, muutub nende ainete koostis geneetilisel tasemel. Seega muutuvad perifeersed kuded paratüreoidhormooni suhtes tundetuks.
Sümptomid
Albrighti sündroomil on kolm peamist sümptomit:
- Kõrge hormonaalse taseme tõttu tekib varajane puberteet. Tüdrukutel võib menstruatsioon alata esimese kümne elukuu jooksul. Kuid see sümptom ei kehti poiste kohta. Neil algab puberteet nagu tavaliselt. Hormonaalne taust suureneb, kui neerupealiste, kilpnäärme ja hüpofüüsi töös ilmnevad talitlushäired. See sümptom võib areneda kahel kujul - mittetäielik ja täielik. Kui patoloogia on täies vormis, algab menstruatsioon liiga vara. Siiski on need valusad ja rohked. Ilmuvad ka sekundaarsed seksuaalomadused, näiteks suurenevad piimanäärmed. Kui patoloogia vorm on puudulik, siis menstruatsioon ei alga. Sageli kaasneb Albrighti tõvega tüdrukutel tsüstide teke munasarjades. Poiste varajast puberteeti iseloomustab peenise suurenemine ja häbemekarvade ilmumine.
- Kiuline osteodüsplaasia. See on patoloogia, mille käigus luukoe deformeerub, mille tõttu luustiku luud on painutatud. Tundub asümmeetriana. Visuaalsed sümptomid on selgroo kõverus ja lonkatus. See patoloogia mõjutab peamiselt torukujulisi luid. Inimese kasv aeglustub. Seda võib täheldada isegi lastel. Kolju luud arenevad ebaühtlaselt, tekivad punnis silmad. Kui puberteet on möödas, peatuvad luumuutused.
- Nahavärvi muutus. Hakkab ilmnema tugev pigmentatsioon. Reitele ja kehale endale tekivad ebaselgete servadega kollakaspruunid laigud. Laigud on selgelt nähtavad alaseljal, ülaseljal, rinnal ja tuharatel. Albrighti sündroomi korral ilmuvad lapse kehale laigud kohe pärast sündi.
Haigus pseudohüpoparatüreoidism näitab spetsiifilise iseloomuga sümptomeid.
Diagnostika
Pseudohüpoparatüreoidismi korral tehakse diagnoos visuaalse läbivaatuse põhjal.
Kasutatakse ka laboratoorseid ja riistvarauuringuid:
- diagnoos tehakse kahe peamise sümptomi ilmnemisel;
- on ette nähtud konsultatsioon geneetikuga;
- tehakse spetsiaalne röntgenuuring;
- teostada laborianalüüs veri hormoonide jaoks;
- vajalik on laste endokrinoloogi konsultatsioon;
- tüdrukuid vaatab läbi lastegünekoloog, poisse uroloog;
- tüdrukutele määratakse alumiste vaagnaelundite ultraheliuuring ja poistele tehakse munandikotti ultraheliuuring;
- vajadusel määratakse endokriinsüsteemi täiendavad uuringud.
Albrighti sündroomi all kannatavaid patsiente peaksid regulaarselt kontrollima sellised spetsialistid nagu endokrinoloog, silmaarst, günekoloog, traumatoloog.
Ravi
Pseudohüpoparatüreoidismi ravitakse kõikehõlmavalt.
Kuna tegemist on geneetilise patoloogiaga, on ravi suunatud ilmnenud sümptomite kõrvaldamisele:
- Määratud hormonaalne ravi. See aitab kaasa endokriinsüsteemi tööle. Patoloogia õigeaegseks arengu vältimiseks on vaja hoolikalt jälgida kolju luude deformatsiooni.
- Kirurgia. Seda meetodit kasutatakse juhul, kui näo luude deformatsioon võib põhjustada kuulmis- ja nägemiskahjustusi.
- Määrake bisfosfonaate sisaldavad valuvaigistid.
Iga Albrighti sündroomi all kannatava patsiendi jaoks valitakse ravimeetod individuaalselt. Patsient peab rangelt järgima kõiki arsti soovitusi ja ettekirjutusi.
Patsiendi kehas jälgitakse perioodiliselt kaltsiumi taset. peal esialgne etapp ravi, viiakse selline uuring läbi kord nädalas. Järk-järgult vähendatakse mõõtmiste sagedust kord kuus. Seda tehakse kuni ettenähtud ravi lõpuni.
Kui diagnoositakse pseudohüpoparatüreoidism, peab patsient järgima ranget dieeti. See seisneb fosforit sisaldavate toodete täielikus menüüst väljajätmises.
Ärahoidmine
Ennetamine on järgmine:
- sellise patoloogiaga inimesed peaksid regulaarselt treenima, et tugevdada lihaseid;
- kui naine soovib rasestuda, siis on vaja konsulteerida günekoloogi ja geneetikuga.
Seda patoloogiat diagnoositakse ühel inimesel miljoni planeedi elaniku kohta. Spetsiifilist ravi kahjuks veel ei ole. On ainult sümptomaatiline ravi. Kuid kui patsient on arsti järelevalve all, on soodsa tulemuse võimalus.
Albrighti sündroom ehk pseudohüpoparatüreoidism on väga haruldane geneetiliselt määratud haigus, mis väljendub kaltsiumi ja fosfori ainevahetuse häiretes ning sellest tulenevalt luusüsteemi patoloogias. Põhisümptomitega kaasneb reeglina ka arengu mahajäämus – nii vaimne kui füüsiline. Haigus esineb sagedamini naistel.
Etioloogia
Teadlased usuvad, et Albrighti sündroomi põhjuseks on paratüreoidhormoonide terved neeru- ja luurakud. Selle põhjuseks on retseptorite defekt. Neerudes on häiritud cAMP (tsükliline aminasiinmofosfaat) süntees, mis on kõrvalkilpnäärme hormooniga seotud metaboolsete protsesside abilüli. Sellised kahjustused tekivad siis, kui kahjustatud on konkreetsed geenid.
Rikkumine võib olla metaboolse ahela mis tahes osas. Mõnel patsiendil on retseptor ise kahjustatud, teistel muutuvad rakumembraani valgud, teistel on haigus seotud cAMP ebapiisava tootmisega. Kõik need juhtumid on seotud patoloogia geneetilise tingimuslikkusega.
Patogenees
Albrighti sündroom on tihedalt seotud kõrvalkilpnäärme füsioloogiaga. Tavaliselt reguleerivad nad kaltsiumi ja fosfori taset veres, kuid selle patoloogilise seisundi korral on nad ükskõiksed nende elementide kõikumise suhtes, mis võib põhjustada sekundaarset hüperparatüreoidismi. Kuid isegi kõrge parahormooni tase ei stimuleeri fosfori ja cAMP kiirendatud väljutamist verest (kuna neerud on selle mõju suhtes puutumata) ja samal ajal kutsuvad esile muutusi luude struktuuris.
Reeglina esineb kõrvalkilpnäärmete kompenseeriv tõus, luude poorsus, tsüstiliste moodustiste tekkimine neis, lupjumised nahas, rasvkoes, lihastes, südames, veresoontes ning sidekestas ja sarvkestas.
Kliinik

Albrighti sündroom avaldub peaaegu samamoodi nagu tõeline hüpoparatüreoidism. Patsientidel tekivad krambid, mis võivad tekkida nii iseseisvalt kui ka pärast kokkupuudet ärritavate teguritega (närvipinge, temperatuurimuutused, raske füüsiline töö jne). Nahas ja nahaaluses rasvas paiknevad kaltsifikatsioonid võivad haavanduda ja põhjustada patsientidele ebamugavusi. Sel juhul otsib patsient abi mitte endokrinoloogilt või geneetikult, vaid kirurgilt või dermatoloogilt.
Sellised inimesed on tavaliselt lühikest kasvu, neil on ümar pundunud nägu, rasvumine ja lühikesed sõrmed. Vale liigeseid täheldatakse seal, kus neid ei tohiks olla, nendes kohtades, kus anatoomiliselt eeldatakse, pole liikuvust, liigeste ja luude konfiguratsiooni rikkumine. Sümptomiteks on ka oksendamine, veri uriinis, katarakt ja muutused hambaemailis.
Lisaks on lapsepõlvest saadik vere madala kaltsiumisisalduse tõttu patsientidel vähenenud intelligentsus ja arengupeetus.
Diagnostika

Albrighti sündroom on haigus, mida on imikutel üsna raske diagnoosida, kuid vanuses 5-10 aastat ilmnevad üsna silmatorkavad ilmingud, mis ei jäta arsti diagnoosis kahtlema. Lastel täheldatakse luusüsteemi hulgi väärarenguid, väheneb kaltsiumi tase veres ning suureneb fosfori ja paratüreoidhormooni tase.
Diagnoosi kinnitamiseks võib teha analüüsi fosfaadi ja cAMP sisalduse kohta uriinis. Selleks süstitakse patsiendile intravenoosselt 200 ühikut parathormooni ja seejärel kogutakse 4 tunni pärast osa uriinist. Kui olulist tõusu ei täheldata, võib see viidata neerukoe resistentsusele paratüreoidhormooni suhtes.
Need märgid on piisavad, et panna Albrighti sündroom. Diagnoosi saab täiendada radiograafiaga. See võimaldab näha konkreetseid muutusi luusüsteemis ja pehmed koed patsient.
Lisaks paratüreoidhormoonile registreerivad selle diagnoosiga inimesed resistentsust ka teistele hormoonidele, mistõttu naistele määratakse Shereshevsky-Turneri sündroomiga diferentsiaaldiagnoos. Nendel kahel haigusel on väline sarnasus ning diagnoosi kontrollimiseks peab arst määrama sugukromatiini analüüsi ning andma saatekirja günekoloogilisele läbivaatusele ja vaagnaelundite ultraheliuuringule.
Ravi ja prognoos

Kui Albrighti sündroomi diagnoos on kindlaks tehtud, tuleb ravi alustada viivitamatult, kuna viivitused süvendavad vaimseid puudujääke. Lastele määratakse kaltsiumipreparaate piisavas koguses päevas, et säilitada normaalne seerumikontsentratsioon. Seejärel lisage sellele D-vitamiini tarbimine annuses, mis ei ületa 100 000 ühikut päevas.
Ravi kohandamiseks on vaja jälgida kaltsiumi taset iga 7 päeva järel esimese kahe nädala jooksul ja seejärel üks kord kuus kuni ravi lõpuni. Kui kaltsiumi optimaalne kontsentratsioon veres on saavutatud, saab seda kontrollida kord kvartalis.
Sekundaarse hüperparatüreoidismi sümptomite kõrvaldamiseks on soovitatav järgida vähendatud fosforisisaldusega dieeti. Kui tuvastatakse teiste hormoonide puudus, viiakse läbi asendus- ja sümptomaatiline ravi.
Kui ravi alustatakse õigeaegselt ja ravi on ratsionaalne, on elu prognoos soodne. Kuid enne rasedust peavad sellise diagnoosiga naised konsulteerima geneetikuga.



