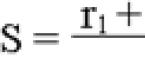Фармацевтическая эквивалентность
Лекарственные препараты фармацевтически эквивалентны, если они содержат одни и те же активные субстанции в одинаковом количестве и в одинаковой лекарственной форме, отвечают требованиям одних и тех же или сходных стандартов и идентичны по силе действия или концентрации активных веществ. Часто, несмотря на одинаковое содержание действующего вещества, дженерический препарат отличается от оригинального по составу вспомогательных веществ
Состав оригинального препарата Вигамокс и дженерического Моксицина в пересчете на 5 мл раствора
- · Вигамокс (28)
- · Моксицин (29)
Действующее веществом оксифлоксацина гидрохлорид 0,02725 г моксифлоксацина гидрохлорид 0, 02725 г
Консервант бензалкония хлорид
Другие вспомогательные вещества натрия хлорид натрия хлорид
кислота борная
кислота хлороводородная и/или натрия гидроксид (для регулировки рН)
вода для инъекций
В состав дженерического моксифлоксацина гидрохлорида входит консервант, оригинальный препарат Вигамокс консерванта не содержит.
Биоэквивалентность
Два лекарственных препарата считаются биоэквивалентными, если они фармацевтически эквивалентны, имеют одинаковую биодоступность и после назначения в одинаковой дозе являются сходными, обеспечивая должную эффективность и безопасность. Под биодоступностью понимается скорость и доля всасывания активного ингредиента или активного компонента лекарства, которое начинает действовать в точке приложения.
В сущности, биоэквивалентность -- это эквивалентность скорости и степени всасывания оригинала и дженерика в одинаковых дозах по концентрации в жидкостях и тканях организма. Достоверность результатов сравнительного исследования биоэквивалентности во многом зависит от соблюдения требований (GМP -- надлежащая клиническая практика) и должно быть независимым, многоцентровым, рандомизированным, контролируемым, длительным.
Если дженерик разрешен к применению в других странах, он регистрируется в РФ по упрощенной схеме (без определения биоэквивалентности). Таким образом, при регистрации зарубежных дженериков в РФ мы в значительной степени доверяем досье, представляемым фармацевтическими компаниями. Такая «доверчивость» в ряде случаев дорого обходится пациентам, т.к. дженерики по своим фармакокинетическим свойствам могут не соответствовать оригинальному препарату. На примере контрольной проверки биоэквивалентности дженериков оригинальному кларитромицину C.N. Nightingale и соавторы сравнили оригинальный препарат кларитромицина с 40 копиями в отношении биоэквивалентности, применив стандарты американской фармакопеи. Исследование показало, что 70% дженериков растворяются значительно медленнее оригинального препарата, что критично для их усвоения. 80% дженериков отличаются от оригинала по количеству действующего начала в одной единице продукта. Количество примесей, не имеющих отношения к действующему началу, в большинстве образцов больше, чем в оригинале. В «лучшем» дженерике их было 2%, в «худшем» -- 32%. Наличие примесей определило выраженность побочных реакций.
С аналогичной ситуацией сталкиваются и офтальмологи. Congdon N.G. и соавторы (2001) по результатам рандомизированного двойного слепого исследования установили преобладание случаев раздражения конъюнктивы и роговицы в связи с местным применением дженерического НПВС -- диклофенака по сравнению с больными, получавшими брендовый препарат.
1. Взаимозаменяемость лекарственных препаратов для медицинского применения определяется в порядке , установленном Правительством Российской Федерации, на основании следующих параметров:
1) эквивалентность (для биоаналоговых (биоподобных) лекарственных препаратов (биоаналогов) - сопоставимость) качественных и количественных характеристик фармацевтических субстанций (использование различных солей, эфиров, комплексов, изомеров, кристаллических форм и других производных одного и того же действующего вещества не является препятствием для взаимозаменяемости лекарственных препаратов, если при проведении исследования биоэквивалентности лекарственного препарата или в случае невозможности проведения этого исследования при проведении исследования терапевтической эквивалентности лекарственного препарата доказано отсутствие клинически значимых различий фармакокинетики и (или) безопасности и эффективности лекарственного препарата для медицинского применения);
2) эквивалентность лекарственной формы (под эквивалентными лекарственными формами понимаются разные лекарственные формы, имеющие одинаковые способ введения и способ применения, обладающие сопоставимыми фармакокинетическими характеристиками и фармакологическим действием и обеспечивающие также достижение необходимого клинического эффекта. Различия лекарственных форм не являются препятствием для их взаимозаменяемости, если при проведении исследования биоэквивалентности лекарственного препарата или в случае невозможности проведения этого исследования при проведении исследования терапевтической эквивалентности лекарственного препарата доказано отсутствие клинически значимых различий фармакокинетики и (или) безопасности и эффективности лекарственного препарата для медицинского применения);
3) эквивалентность или сопоставимость состава вспомогательных веществ лекарственного препарата для медицинского применения (различия состава вспомогательных веществ лекарственного препарата для медицинского применения не являются препятствием для их взаимозаменяемости, если при проведении исследования биоэквивалентности лекарственного препарата для медицинского применения или в случае невозможности проведения этого исследования при проведении исследования терапевтической эквивалентности лекарственного препарата для медицинского применения доказано отсутствие клинически значимых различий фармакокинетики и (или) безопасности и эффективности лекарственного препарата для медицинского применения. При этом различия состава вспомогательных веществ не должны приводить к риску возникновения серьезных нежелательных реакций у отдельных групп пациентов или повышения частоты их возникновения);
4) идентичность способа введения и применения;
5) отсутствие клинически значимых различий при проведении исследования биоэквивалентности лекарственного препарата или в случае невозможности проведения этого исследования отсутствие клинически значимых различий показателей безопасности и эффективности лекарственного препарата при проведении исследования терапевтической эквивалентности. Данный параметр не применяется в отношении воспроизведенных лекарственных препаратов, указанных в части 10 статьи 18 настоящего Федерального закона. В отношении биоаналоговых (биоподобных) лекарственных препаратов (биоаналогов) данные об отсутствии клинически значимых различий безопасности, эффективности и иммуногенности лекарственного препарата по результатам проведения клинических исследований предоставляются в порядке, установленном настоящей частью;
6) соответствие производителя лекарственного средства требованиям надлежащей производственной практики.
2. Сравнение параметров регистрируемых лекарственных препаратов для медицинского применения осуществляется комиссией экспертов экспертного учреждения при проведении экспертизы таких лекарственных препаратов в процессе их государственной регистрации. Выводы экспертов о взаимозаменяемости или невзаимозаменяемости лекарственных препаратов для медицинского применения, сделанные в результате этого сравнения, оформляются в виде приложения к заключению экспертов по форме , утвержденной уполномоченным федеральным органом исполнительной власти.
3. Положения настоящей статьи не распространяются на референтные лекарственные препараты, лекарственные растительные препараты, гомеопатические лекарственные препараты и лекарственные препараты, которые разрешены для медицинского применения в Российской Федерации более двадцати лет и в отношении которых невозможно проведение исследования их биоэквивалентности.
Проблема взаимозаменяемости лекарственных средств (ЛС) в настоящее время обсуждается довольно активно на самых разных уровнях и в нашей стране, и за рубежом. Она имеет как чисто терапевтическую, так и довольно актуальную экономическую подоплеку. Следует отметить, что в нашей стране пока нет четкого определения взаимозаменяемых ЛС. Трактовка этого понятия очень сильно зависит от мнения разных специалистов и опыта применения ими ЛС. Совершенно разное отношение к этому термину может возникнуть при решении чисто терапевтических задач и задач, связанных с закупкой ЛС по государственным контрактам.
Тем не менее мы сконцентрируемся на некоторых конкретных аспектах проблемы взаимозаменяемости. С терапевтической точки зрения очевидно, что два разных ЛС (т.е. содержащих два разных действующих вещества) одной фармакологической группы могут заменять друг друга при условии, что они имеют, например, одинаковый механизм действия (скажем, блокада одних и тех же рецепторов) и что при необходимости будет проведена соответствующая корректировка доз и кратности приема. И в этом смысле эти два ЛС для врача будут взаимозаменяемы: сейчас нет в продаже одного, и он назначит другое. Это так называемая терапевтическая замена, но понятию взаимозаменяемости она не тождественна. В настоящее время этот термин касается замены оригинального ЛС на дженерик или замены одного дженерика на другой с тем же действующим веществом. В этом, по крайней мере, уже имеется определенная договоренность . А вот какие оригинальные препараты и дженерики следует считать взаимозаменяемыми и какие проблемы стоят на пути решения этого вопроса, мы и обсудим в данной статье.
Проблема эквивалентности воспроизведенных ЛС (дженериков) стоит достаточно остро ввиду большого количества препаратов различных производителей на рынке. Этот вопрос является актуальным во всем мире, но в нашей стране в последние два десятилетия он приобрел особую значимость, поскольку мы оказались не готовы к огромному потоку ЛС. Наша регуляторная система, научная база и все понятия были адаптированы для рынка, страдающего от дефицита ЛС. Теперь же ситуация изменилась кардинально. Российский фармацевтический рынок насыщен большим количеством препаратов. По некоторым действующим веществам (парацетамол, ацетилсалициловая кислота, диклофенак натрия, метамизол натрий, эналаприла малеат, ципрофлоксацина гидрохлорид и др.) еще 10 лет назад было зарегистрировано несколько сотен препаратов разных производителей (с учетом разных лекарственных форм и дозировок). Сейчас число соответствующих препаратов немного снизилось, но это проблему дженериков ни в коей мере не снимает.
В чем же заключается вопрос? Ведь избыток товара должен способствовать конкуренции, борьбе за клиента путем повышения качества продукции и снижения ее стоимости. И именно с этого места в дальнейшем в данной статье мы будем стараться избегать слова «товар», поскольку говорить будем о ЛС, т.е. о продукции, обладающей уникальными свойствами, для которой первоочередными критериями являются качество, эффективность и безопасность. Все же остальное является следствием – и цена препаратов, и здоровье, которое мы рискуем потерять, применяя ЛС, не удовлетворяющие современным требованиям.
Требования к ЛС
Начиная обсуждение подходов к оценке взаимозаменяемости ЛС, стоит привести выдержку из совместного заявления Международной фармацевтической федерации (FIP, www.fip.org) и Международной федерации фармацевтических производителей и ассоциаций (IFPMA, www.ifpma.org), принятого в 1999 г., в котором, в частности, говорится, что замена оригинального ЛС на воспроизведенное «должна проводиться только в том случае, когда имеется соответствие принятым международным стандартам, включая биоэквивалентность, с целью гарантирования качества всех препаратов на рынке» .
В этом же документе указывается: «Все правительства должны предпринять шаги к обеспечению качества, безопасности и эффективности всех ЛС, доступных в соответствующих государствах, согласно принятым международным стандартам. Это относится как к оригинальным, так и к воспроизведенным ЛС, к частному и государственному секторам и к импортируемой и производимой на местном рынке продукции».
Таким образом, к любому ЛС предъявляются три требования: эффективность, безопасность и качество. Такой подход к оценке ЛС в настоящее время принят во всем мире, в т.ч. и в нашей стране.
Категории «эффективность» и «безопасность» относятся к медико-биологическим вопросам, а «качество» является чисто фармацевтической проблемой и отражает соответствие ЛС требованиям нормативной документации по показателям подлинности (т.е. идентичности содержимого упаковки), чистоты (по содержанию примесей) и количественного содержания действующего вещества (или веществ в случае комбинированного препарата). С обеспечением и контролем качества ЛС неразрывно связаны правила надлежащей производственной практики (GMP).
Оригинальные и воспроизведенные ЛС. Основные термины
При обсуждении вопросов взаимозаменяемости ЛС следует определиться с основными терминами. В Федеральном законе №61-ФЗ «Об обращении лекарственных средств» дается следующее определение: оригинальное ЛС – ЛС, содержащее впервые полученную фармацевтическую субстанцию или новую комбинацию фармацевтических субстанций, эффективность и безопасность которых подтверждены результатами доклинических исследований ЛС и клинических исследований лекарственных препаратов . В зарубежной литературе можно встретить соответствующие термины «innovator product», «branded product». В документах ВОЗ «innovator pharmaceutical product» – это продукт, впервые разрешенный к применению на основе документации о его качестве, безопасности и эффективности.
Организация, разработавшая оригинальное ЛС, получает на него патентную защиту на длительный период (до 20 лет и более), позволяющую компенсировать высокие затраты на разработку средства и его продвижение на рынке, а также получить дополнительный доход. Оригинальные ЛС имеют высокую стоимость во время действия патентной защиты и более высокую стоимость по сравнению с дженериками после ее окончания.
Это обстоятельство вызывает постоянную критику. Утверждается, что декларируемые ведущими производителями расходы на разработку нового ЛС (т.е. новой молекулы) явно завышены, как неоправданно завышена и стоимость оригинальных средств после окончания действия патента.
Но не стоит забывать, что, во-первых, современные ЛС имеют более тонкие, направленные, избирательные механизмы действия, чем средства, например, из тех же фармакологических групп, но разработанные несколько десятилетий назад. Поэтому даже при современном скрининге активных веществ, позволяющем в ряде случаев существенно сократить время поиска новой активной молекулы, период поиска, умноженный на стоимость используемых современных высоких технологий, оборачивается существенными финансовыми затратами.
Во-вторых, мы не устанем повторять, что основным локомотивом развития фармацевтической отрасли являются производители инновационных ЛС. Следует помнить, что прибыль, получаемая при реализации оригинальных препаратов, направляется в конечном счете, на разработку инновационных ЛС.
Воспроизведенное ЛС – ЛС, содержащее такую же фармацевтическую субстанцию или комбинацию таких же фармацевтических субстанций в такой же лекарственной форме, что и оригинальное ЛС, и поступившее в обращение после поступления в обращение оригинального ЛС . В отношении воспроизведенных ЛС применяют также термин «дженерик» или «генерик» (generic product).
Следует отметить, что в документах ВОЗ (www.who.int) в связи с различной трактовкой в разных странах термина «дженерики» рекомендуется использовать термин «мультиисточниковые лекарственные препараты» (multisource pharmaceutical products). ВОЗ определяет их как фармацевтически эквивалентные или фармацевтически альтернативные препараты, которые могут быть или не быть терапевтически эквивалентными . Термин «мультиисточниковый», хоть и не очень благозвучный и не уживающийся в отечественной терминологии, но конкретизирует понимание того, что воспроизведенные ЛС производятся из фармацевтических субстанций и вспомогательных веществ, имеющих самое разное происхождение. То есть собираются, как конструктор, – из деталей разных производителей. Поскольку теоретически поставщики могут легко меняться, то это уже является поводом для критики со стороны производителей оригинальных ЛС. Действительно, такой подход в общем случае затрудняет обеспечение однородного качества, а также соответствия требованиям эффективности и безопасности.
В этом отношении интересно определение дженерика, которое дается в Директиве 2001/83/EC Европейского парламента, определяющей требования к лекарственным средствам: генерический лекарственный препарат (generic medicinal product) означает лекарственный препарат, имеющий такой же качественный и количественный состав в отношении активной субстанции и ту же лекарственную форму, что и препарат сравнения, и биоэквивалентность которого была доказана путем проведения исследований биодоступности по отношению к препарату сравнения. То есть страны Евросоюза теоретически не допускают наличие на рынке дженериков с проблемами биоэквивалентности, хотя, конечно, это вовсе не исключает соответствующую проблему.
Согласно определению ВОЗ лекарственные препараты являются фармацевтически эквивалентными, если они содержат одинаковое количество одной и той же активной субстанции (субстанций) в одной и той же лекарственной форме, отвечают сопоставимым стандартам качества и предназначены для одного пути введения . Аналогичное, но более конкретное и удобное с практической точки зрения определение дает FDA США (www.fda.gov): препараты считаются фармацевтически эквивалентными, если содержат (1) одинаковые активные ингредиенты (2) с одинаковой дозировкой или концентрацией (3) в одинаковой лекарственной форме, (4) предназначены для одного пути введения и (5) соответствуют фармакопейным или другим применяемым стандартам по количественному содержанию, чистоте и подлинности .
ВОЗ указывает, что лекарственные препараты являются фармацевтически альтернативными, если они содержат одинаковое количество одного и того же активного начала, но различаются по лекарственной форме (например, таблетки и капсулы) и/или по химической форме (различные соли, эфиры). Фармацевтически альтернативные препараты предназначены для одного пути введения . В определении фармацевтически альтернативных препаратов примерно такой же подход и у FDA .
Необходимо обратить внимание, что вплоть до настоящего времени в литературе, в интернете, в устной речи можно встретить достаточно вольное употребление терминов «синонимы ЛС», «аналоги лекарств», «заменители лекарств» и т.п. Мы хотим предостеречь специалистов от использования данной терминологии без четкого обозначения контекста, соответствующего вышеуказанным подходам.
Например, термин «синонимы ЛС» неправильно продолжают использовать по отношению к дженерикам. Это было бы хоть в некоторой степени оправданно, если бы не существовало проблемы терапевтической эквивалентности, взаимозаменяемости. Поэтому синонимами не могут быть разные торговые наименования препаратов. Но для субстанций это могут быть, например: метамизол натрий как международное непатентованное название (МНН) и анальгин как отечественное фармакопейное название, парацетамол как МНН и ацетаминофен как название, принятое в США (USAN).
Биоэквивалентность дженериков
Одинаковые действующее вещество, дозировка, лекарственная форма и путь введения «сближают» лекарственные препараты разных производителей, но это не означает, что эффективность дженериков будет также эквивалентна, т.к. разница в параметрах качества действующей субстанции (например, полиморфизм), в составе вспомогательных веществ и/или производственном процессе может привести к различиям в эффективности данных препаратов. Этот факт давно известен и обсуждается с середины прошлого столетия .
В качестве небольшого отступления отметим, что довольно часто обсуждается вопрос влияния примесей на эффективность и безопасность дженериков. По этому поводу можно сказать, что при соблюдении норм чистоты, заложенных в единый государственный фармакопейный стандарт качества ЛС, вопрос влияния примесей в общем случае снимается. Другое дело, когда такого стандарта качества (т.е. Государственной фармакопеи) в полноценном виде фактически нет, а существующая нормативная документация производителей (ФСП и НД) весьма разнообразна. В этом случае вопрос о примесях так же, как и о других параметрах качества, остается открытым, что оказывает косвенное влияние на эффективность и безопасность препаратов.
Итак, в связи с неоднородностью дженериков возникло понятие биологической эквивалентности. Согласно документам ВОЗ, два лекарственных препарата являются биоэквивалентными, если они фармацевтически эквивалентны или являются фармацевтически альтернативными, а их биодоступность с точки зрения максимальной концентрации в плазме крови (CMAX), времени достижения этой концентрации (TMAX) и площади под фармакокинетической кривой (AUC) после применения в одинаковой молярной дозе при одинаковых условиях сходна до такой степени, в которой их эффекты являются по существу одинаковыми . Примерно такой же подход у Европейского агентства по лекарственным средствам (EMA, www.ema.europa.eu) и у FDA .
ВОЗ и FDA рекомендуют определять биоэквивалентность с использованием следующих испытаний in vivo и in vitro:
— сравнительные фармакокинетические испытания на людях (изучение концентрационного профиля ЛС или его метаболитов в биологических жидкостях);
— сравнительные фармакодинамические испытания на людях (изучение эффектов, вызываемых ЛС);
— сравнительные клинические испытания;
— сравнительные исследования in vitro (например, тест «растворение»).
Слово «сравнительные» означает, что все вышеперечисленные исследования проводятся путем сопоставления соответствующих параметров у испытуемого препарата и препарата сравнения.
ВОЗ публикует рекомендации по выбору препарата сравнения для жизненно необходимых ЛС. Эти рекомендации включают, в частности, для каждого МНН торговое название оригинального лекарственного препарата, который рекомендуется использовать в качестве препарата сравнения при установлении взаимозаменяемости дженериков. При этом также приводятся соответствующие лекарственные формы и дозировки.
В США выбор препарата сравнения проводится на основе данных, представленных в так называемой Оранжевой книге , о которой мы будем говорить ниже. В ней также дается указание на конкретный лекарственный препарат в определенной лекарственной форме, определенной дозировке и конкретного производителя. Препараты, которые можно использовать для сравнения с ними дженериков, имеют пометку в соответствующей графе. Имеется также отечественная документация , в которой приводятся указания по проведению фармакокинетических исследований и теста «растворение» для оценки биоэквивалентности ЛС.
Терапевтическая эквивалентность и взаимозаменяемость
И наконец, самое важное понятие – терапевтическая эквивалентность. Важное, потому что ближе всего стоит к пониманию того, какие препараты могут быть взаимозаменяемы. Действительно, согласно определению ВОЗ, взаимозаменяемый лекарственный препарат – это препарат, который является терапевтически эквивалентным при сопоставлении с препаратом сравнения и на который препарат сравнения можно заменить в клинической практике . Такая же позиция отражена и в документах FDA .
Мы также будем придерживаться подхода, при котором терапевтически эквивалентные препараты будут являться взаимозаменяемыми. Остается только решить вопрос: какие препараты считать терапевтически эквивалентными?
Критерии терапевтической эквивалентности
Начнем с определения ВОЗ: «два лекарственных препарата являются терапевтически эквивалентными, если они фармацевтически эквивалентны или являются фармацевтически альтернативными и после их применения в одной молярной дозе их эффективность и безопасность являются по существу одинаковыми, когда они применяются одним путем при условиях, описанных в инструкции» .
Теперь приведем определение терапевтической эквивалентности, которое дается FDA: «лекарственные препараты считаются терапевтически эквивалентными, только если они являются фармацевтически эквивалентными и имеют одинаковые клинические эффекты и профиль безопасности и когда они применяются при условиях, описанных в инструкции» . То есть, в отличие от ВОЗ, FDA рассматривает в качестве терапевтических эквивалентов только фармацевтически эквивалентные препараты. Таким образом, с точки зрения FDA капсулы и таблетки, например, даже в одной дозе не будут терапевтически эквивалентны. Более того, FDA после общего определения довольно конкретно описывает все условия терапевтической эквивалентности.
1. Лекарственные препараты должны быть разрешены к применению как эффективные и безопасные.
2. Препараты должны являться фармацевтически эквивалентными.
3. Препараты должны являться биоэквивалентными, т.е. должны соблюдаться условия:
— для них не известны существующие или отсутствуют потенциальные проблемы с биоэквивалентностью, и они отвечают требованиям соответствующего стандарта при проведении испытаний in vitro или
— для них известны существующие или возможны потенциальные проблемы с биоэквивалентностью, но было показано, что они соответствуют требованиям подходящего стандарта биоэквивалентности.
4. Препараты должны иметь надлежащую инструкцию.
5. Препараты должны производиться в соответствии с требованиями Надлежащей производственной практики (т.е. GMP).
FDA издает важный документ, имеющий официальное название «Approved Drug Products with Therapeutic Equivalence Evaluations», которое можно перевести примерно как «Разрешенные к применению лекарственные препараты с указанием их терапевтической эквивалентности» . Коротко этот документ принято называть Оранжевой книгой.
Собственно, указанные выше подходы к оценке терапевтической эквивалентности препаратов конкретных производителей и отражены FDA в этом издании. Специалисты FDA указывают, что использование соответствующих кодов терапевтической эквивалентности может служить ориентиром при замене одного препарата на другой и помочь, в частности, в снижении стоимости лечения. Необходимо также помнить, что основная ценность Оранжевой книги FDA состоит в том, что она имеет электронную версию, причем обновляемую ежедневно.
Следует отметить, что в настоящее время в отечественной терминологии сложилась ситуация, когда под испытанием биоэквивалентности подразумевают только изучение концентрационного профиля лекарственного вещества, т.е. фармакокинетические исследования. И отечественные специалисты считают, что нельзя отождествлять биоэквивалентность и терапевтическую эквивалентность, поскольку последнюю можно подтвердить только путем проведения полноценных клинических исследований по протоколу. Зарубежная же документация подразумевает либо прямо указывает на тождественность данных понятий . Поэтому в Оранжевой книге FDA отметка о терапевтической эквивалентности может быть поставлена на основании положительного результата при изучении биоэквивалентности фармакокинетическим подходом. Самодостаточность таких исследований в большинстве случаев обусловлена тем, что концентрационный профиль лекарственного вещества в плазме крови соответствует таковому в месте действия.
Некоторые критерии терапевтической эквивалентности, применяемые FDA, мы также обсудим ниже. Но в целом считаем, что подход FDA к решению проблемы взаимозаменяемости лекарственных препаратов является вполне обоснованным и наиболее проработанным. Но с обязательным выполнением всех вышеуказанных условий. Подчеркнем, что коды терапевтической эквивалентности могут служить ориентиром в направлениях «эффективность» и «безопасность», если решена проблема в направлении «качество».
Терапевтическая эквивалентность и GMP
Устанавливая критерии терапевтической эквивалентности (т.е. взаимозаменяемости) лекарственных препаратов, FDA указывает на необходимость соответствия их производства требованиям GMP. Это, безусловно, очень важно. Действительно, если ЛС производятся не по стандартам GMP, они не могут быть однородными от серии к серии. И это сказывается на всех параметрах препарата: на качестве, эффективности и безопасности. Поэтому доказанная терапевтическая эквивалентность для одной серии такой продукции вовсе не будет означать, что вся продукция в последующем будет соответствовать необходимым стандартам биоэквивалентности.
При этом надо понимать, что государственный контроль качества не решает проблему в целом, даже если это было бы возможно сделать для всех выпускаемых и импортируемых ЛС. Поэтому в настоящее время во всем мире и в нашей стране речь идет о смещении акцента с контроля качества ЛС на обеспечение качества. Ранее мы уже неоднократно указывали на то, что у производителя на выходе имеются качественные лекарства тогда, когда они получаются таковыми в процессе производства, а не потому, что хорошо отбраковываются некачественные. Следование такому принципу влечет за собой полное соответствие предприятия требованиям GMP (и даже больше), введение института уполномоченных лиц, привлечение к работе исключительно квалифицированного персонала, сокращение острейшего дефицита специалистов (технологов, аналитиков и др.) и многое др.
Здесь мы также добавим, что большую роль в обеспечении качества ЛС играют собственные ресурсы производителя в области научных исследований и разработок. И чем эти ресурсы более развиты и более тесно взаимодействуют с подразделениями производства, контроля и обеспечения качества, тем в большей степени можно быть уверенным в качестве соответствующей продукции.
Установление эквивалентности без проведения исследований
Исходная позиция такова, что если ЛС предназначено для системного применения (лекарственное вещество определяется в системном кровотоке), то необходимы фармакокинетические исследования биоэквивалентности. Если же действие препарата не обусловлено появлением активного вещества в системном кровотоке или его определить там затруднительно имеющимися аналитическими методами, то тогда необходимы фармакодинамические или даже полноценные клинические исследования .
Но возникает вопрос о сравнении, например, инъекционных средств. Если это уже готовые растворы, являющиеся фармацевтически эквивалентными препаратами и предназначенные для внутривенного введения, то как установить их терапевтическую эквивалентность и надо ли это вообще делать?
Согласно ВОЗ , воспроизведенные ЛС могут считаться терапевтически эквивалентными без дополнительных исследований в следующих случаях.
1. Лекарственные препараты предназначены для парентерального (внутривенного, подкожного или внутримышечного) введения и представляют собой водные растворы одной и той же активной субстанции в той же молярной концентрации, а также содержат те же самые или подобные вспомогательные вещества в примерно одинаковых концентрациях. Определенные вспомогательные вещества (например, создающие буферную среду, а также консерванты и антиоксиданты) могут отличаться при следующем условии: если может быть продемонстрировано, что это не влияет на безопасность и/или эффективность лекарственного препарата.
2. Лекарственные препараты представляют собой растворы для перорального применения (например, сиропы, эликсиры, настойки), содержащие одно и то же действующее вещество в одной и той же молярной концентрации и, по существу, одни и те же вспомогательные вещества в сравнимых концентрациях. При этом пристальное внимание следует уделить рассмотрению тех вспомогательных веществ, которые влияют на всасывание и стабильность действующего вещества в желудочно-кишечном тракте.
3. Лекарственные препараты представляют собой порошки для приготовления растворов, и получаемые растворы должны соответствовать критериям, указанным в пунктах 1 или 2.
4. Лекарственные препараты являются газами.
5. Ушные и глазные лекарственные препараты, являющиеся водными растворами и содержащие одну и ту же активную субстанцию (субстанции) в одной и той же молярной концентрации и, по существу, одни и те же вспомогательные вещества в сравнимых концентрациях. Определенные вспомогательные вещества (например, консерванты, вещества, создающие буферную среду, корректирующие осмотические свойства и вязкость) могут отличаться при условии, что их использование не должно повлиять на безопасность и/или эффективность препарата.
6. Лекарственные препараты для местного применения, являющиеся водными растворами и содержащие одну и ту же активную субстанцию (субстанции) в одной и той же молярной концентрации и, по существу, одни и те же вспомогательные вещества в сравнимых концентрациях.
7. Лекарственные препараты, являющиеся водными растворами для использования в виде ингаляций в небулайзере или в качестве назальных спреев, предназначенные для применения с использованием, по существу, одинаковых устройств и содержащие одну и ту же активную субстанцию (субстанции) в одной и той же концентрации и, по существу, одни и те же вспомогательные вещества в сравнимых концентрациях. Лекарственные препараты могут содержать разные вспомогательные вещества при условии, что их использование не должно повлиять на безопасность и/или эффективность препаратов.
Таким образом, вышеуказанные ситуации снимают необходимость проведения соответствующих исследований. А отсутствие влияния отличающихся по составу вспомогательных веществ в указанных случаях заявитель, очевидно, должен продемонстрировать путем предоставления дополнительной информации. В целом же надо понимать, что гарантией терапевтической эквивалентности лекарственного препарата в описанных случаях будет соответствие его качества требованиям нормативной документации.
Отдельные проблемы, требующие внимания
При использовании ЛС, при их закупке по государственным контрактам хотелось бы иметь четкое руководство, в котором было бы прописано, какие препараты являются взаимозаменяемыми. Таким руководством мог бы быть аналог Оранжевой книги. Но при его формировании и при установлении соответствующих правил требуется учесть массу специфических моментов, кроме вышеописанных общих правил. Остановимся на некоторых частных проблемах.
МНН. Известно, что размещение заказа на поставку ЛС должно осуществляться по МНН . Исключение составляют инсулины и циклоспорин, для которых это можно делать по торговым наименованиям. Но необходимо отметить, что МНН (или другое название, если МНН отсутствует) не может служить единственным ориентиром.
Во-первых, препараты с одним действующим веществом (МНН) могут различаться по другим характеристикам (дозировка, лекарственная форма, способ применения), что в общем случае исключает их взаимозаменяемость.
Во-вторых, при формальном подходе есть вероятность совершить ошибку, поскольку ВОЗ присваивает МНН (и это учтено в Государственном реестре ЛС РФ) обычно для кислот и оснований (если таковые существуют). Для солей, эфиров и других производных от основной структуры МНН могут быть присвоены только в том случае, если такие производные являются единственно возможным вариантом. Например, присвоено МНН для соли – метамизол натрий. И сделано это по той причине, что в виде кислоты метамизол не существует из-за неустойчивости, но это редкий случай. При принятии решения ошибка может заключаться в том, что для одного МНН реально могут существовать разные действующие вещества, например: ципрофлоксацин (в виде основания) и ципрофлоксацина гидрохлорид (соль), гидрокортизон (основная структура) и гидрокортизона ацетат (сложный эфир – производное от базовой структуры). Как мы указывали выше, препараты, содержащие разные соли и эфиры, являются фармацевтически альтернативными. Специалисты FDA такие препараты не считают терапевтически эквивалентными, и с этим необходимо согласиться, потому что в общем случае химические модификации действующих веществ преследуют цели изменения растворимости, стабильности, кристалличности, размеров частиц, биодоступности и др., что, в конечном счете, влияет на безопасность и эффективность препарата.
Замена лекарственной формы. Радикальная смена лекарственной формы может дать отрицательный эффект вообще. Например, ЛС для парентерального и перорального введения не эквивалентны и не взаимозаменяемы. Более того, пациент может быть без сознания, и ему требуются инъекции. Может требоваться препарат для ректального введения ввиду того, что пациент не может глотать. Препараты для местного и системного применения, даже если они применяются для лечения одной патологии, имеют совершенно разную эффективность (системные эффективнее).
Известно, что ЛС, имеющие разные пути введения, вообще могут применяться по разным показаниям. Хрестоматийный пример – фенотерол. Препарат Беротек, раствор или аэрозоль для ингаляций – бронходилататор (например, для купирования приступов бронхиальной астмы), а Партусистен, таблетки или концентрат для инфузий – токолитик (предотвращение преждевременных родов).
Здесь также необходимо отметить, что количество единиц лекарственной формы в упаковке тоже может иметь значение. Например, противозачаточные таблетки. В одной упаковке – 21 таблетка. В другой – 28 таблеток, из которых 7 – плацебо. FDA такие препараты не считает терапевтическими эквивалентами.
Фармацевтически альтернативными препаратами являются порошки для инъекций, подлежащие растворению, концентрированные растворы для инъекций, подлежащие разведению, и готовые растворы для инъекций, не требующие подобных предварительных манипуляций. Такие препараты, согласно FDA, также не являются терапевтически эквивалентными.
Комбинированные препараты. Проблемой является замена комбинированного препарата на монопрепараты, содержащие те же действующие вещества и в тех же дозировках, что и в комбинации. Например, в «Обзоре практики рассмотрения жалоб на действия (бездействие) заказчика, уполномоченного органа, специализированной организации, аукционной комиссии при проведении торгов в сфере здравоохранения в соответствии с положениями Федерального закона от 21 июля 2005 г. №94-ФЗ» (подготовлен Управлением контроля размещения государственного заказа ФАС России, июль 2011 г.) указано, что «при порядке формирования Заказчиком лотов на закупку антивирусных препаратов: комбинированных и монопрепаратов, предназначенных для лечения лиц, инфицированных вирусом иммунодефицита человека, необходимо учитывать, что комбинированные и монопрепараты в той же комбинации в виде 2 или 3 таблеток являются взаимозаменяемыми» . Если принять, что это допустимо, то решение этой проблемы должно быть конкретным, т.е. приниматься для каждого препарата на основе научных данных с учетом вышеуказанных критериев терапевтической эквивалентности.
Dura lex…
Деятельность, связанная с ЛС, регулируется законодательно. Это касается в т.ч. и закупок ЛС по государственным контрактам. Например, согласно Федеральному закону №94-ФЗ (ч. 3.1 ст. 34), «Документация об аукционе не может содержать указание на знаки обслуживания, фирменные наименования, патенты, полезные модели, промышленные образцы, наименование места происхождения товара или наименование производителя, а также требования к товару, информации, работам, услугам, если такие требования влекут за собой ограничение количества участников размещения заказа» .
Но, как отмечают специалисты компании «Гарант» , «при этом необходимо помнить, что целью размещения заказа является удовлетворение потребностей заказчика (ст. 3 Закона №94-ФЗ). Как отмечали в связи с этим суды, в зависимости от своих потребностей заказчик должен установить конкретные требования к качеству, к функциональным характеристикам (потребительским свойствам) товара, к размерам, упаковке, т.е. потребности заказчика являются определяющим фактором при установлении им соответствующих требований (постановление ФАС Западно-Сибирского округа от 07.09.2010 по делу №А03-2442/2010)». А вопрос правомерности указания конкретных требований к лекарственному препарату, по нашему мнению, должен решаться с учетом медицинских аспектов, которые рассмотрены выше.
Известна, например, ситуация, сложившаяся в 2010 г. при закупке препарата золедроновой кислоты. При размещении заказа Министерство обороны РФ конкретизировало лекарственную форму и концентрацию препарата таким образом, что это фактически указывало на препарат конкретного производителя – раствор для инфузий 5 мг/100 мл выпускается только под названием Акласта и производится компанией Novartis. В данном случае ФАС было принято решение о нарушении, которое ограничивает количество участников размещения заказов, и заказчику было выдано соответствующее предписание. Решение ФАС было обжаловано Минобороны в арбитражном суде двух инстанций, но оставлено в силе. Возражений со стороны производителя ЛС в данном случае не поступило, т.е. он согласился с тем, что лиофилизат или концентрат для приготовления раствора для инфузий, которые также доступны на рынке, в данном случае будут эквивалентной заменой готовому раствору, хотя, согласно требованиям FDA, это не так.
А вот другая, казалось бы, аналогичная ситуация, по которой в итоге было принято иное решение. В 2010 г. Курганская областная клиническая больница разместила заказ на приобретение препарата, содержащего доцетаксел. При этом в аукционную документацию были включены дополнительные требования: 1) концентрат для приготовления раствора для инфузий 20 мг, объем наполнения флакона 24,4 мг/0,61 мл в комплекте с растворителем 1,98 мл во флаконе №1; 2) концентрат для приготовления раствора для инфузий 80 мг, объем наполнения флакона 94,4 мг/2,36 мл в комплекте с растворителем 7,33 мл во флаконе №1. Такая детализация, по сути, опять указывала на конкретный препарат – Таксотер. Причем заказчик этого и не скрывал: согласно закону №94-ФЗ, в аукционной документации он указал не только МНН, но и торговое название с обязательными словами «или эквивалент». ФАС тем не менее было усмотрено нарушение, поскольку эквивалента с такой формой выпуска фактически нет на рынке, но есть другие препараты, которые могли бы быть заменой. Однако при рассмотрении дела в арбитражном суде решение ФАС было признано недействительным.
Совершенно неожиданный поворот, который даже специалисту, на первый взгляд, кажется странным. Но научное обоснование, на которое ориентировался суд, было следующим. Цитата: «… оспариваемое решение УФАС по Курганской области является незаконным в связи со следующим: заинтересованным лицом не учтено, что указанное в документации об аукционе требование к объёму наполнения флакона — 24,4 мг/0,61 мл и 94,4 мг/2,36 мл является единственно возможным наполнением для обеспечения эффективного применения препарата, необходимого Заказчику, который полагает, что именно такое наполнение обеспечивает надлежащую дозировку с учетом адгезии (оседание на стенках сосуда) препарата и позволяет компенсировать потери жидкости при приготовлении предварительно смешанного раствора, считает, что избыток препарата во флаконе гарантирует после разведения его содержимого нужную дозу 20 мг, указанную на этикетке флакона. Иное наполнение флакона в результате адгезии приведет к действительной дозировке, которая будет ниже необходимой, в результате при применении препарата терапевтического эффекта не будет.
Это же касается и концентрата для приготовления инфузий 80 мг. Настаивает, что только объем наполнения флакона, поставляемого Заявителем и указанный в документации об аукционе (24,4 мг/0,61 мл и 94,4 мг/2,36 мл), позволяет обеспечить точную дозировку без дополнительных измерений, в то время как флакон иного объема создает значительный риск ошибочной дозировки. Таким образом, указание в документации об аукционе на объем наполнения флакона в действительности имело целью обеспечить поставку препарата в том виде, в котором возможно обеспечить его эффективное применение при лечении пациентов с раковыми заболеваниями, данное обстоятельство не было учтено УФАС по Курганской области при вынесении оспариваемого Решения.».
Приведенные примеры еще раз указывают на то, что даже при наличии неких общих ориентиров каждый конкретный случай требует отдельного рассмотрения. К решению данной проблемы может приблизить некий аналог Оранжевой книги, в котором можно указать, что один препарат не имеет взаимозаменяемых препаратов, а другой имеет. Но при этом следует понимать, что это огромная работа. Фактически это научный труд, основанный на доказательствах.
Заключение
При рассмотрении вопросов взаимозаменяемости ЛС следует исходить из того, что воспроизведенные ЛС, содержащие одно и то же действующее лекарственное вещество, не являются терапевтически эквивалентными, а значит, не являются взаимозаменяемыми. Их терапевтическую эквивалентность необходимо доказывать для каждого препарата каждого производителя. Эти доказательства должны строиться на научно обоснованных данных, а принятие конкретного решения о замене препарата должно быть обусловлено медицинской спецификой и может основываться на информации, представленной в соответствующем руководстве.
Еще раз обращаем внимание на тот факт, что при нерешенном вопросе в отношении качества выпускаемых ЛС, в т.ч. в отношении Государственной фармакопеи, принятие решений, связанных с взаимозаменяемостью препаратов, остается затруднительным.
Литература
1. Биологическая доступность лекарственных средств: принципы и проблемы. Докл. Науч. группы ВОЗ №536. – Женева: ВОЗ, 1975.
2. ГАРАНТ ЭКСПЕРТ: ГАРАНТ-Максимум (электронный ресурс).
3. Оценка биоэквивалентности лекарственных средств. Методические указания. – М.: ФГУ НЦ ЭСМП, 2008.
4. Письмо Минэкономразвития России, Минздравсоцразвития России и Федеральной антимонопольной службы РФ от 31.10.2007 №16811-АП/Д04, 8035-ВС и /ИА/20555.
5. Тенцова А.И., Ажгихин И.С. Лекарственная форма и терапевтическая эффективность лекарств. — М.: Медицина, 1974. – 336 с.
6. Федеральный закон № 61-ФЗ от 12 апреля 2010 г. «Об обращении лекарственных средств».
7. Федеральный закон № 94-ФЗ от 21 июля 2005 г. «О размещении заказов на поставки товаров, выполнение работ, оказание услуг для государственных и муниципальных нужд».
8. Холодов Л.Е., Яковлев В.П. Клиническая фармакокинетика. – М.: Медицина, 1985. – 464 с.
9. Approved Drug Products with Therapeutic Equivalence Evaluations, 31st ed., FDA, 2011.
10. CPMP/EWP/QWP/1401/98 Rev. 1/ Corr.: Guideline on the Investigation of Bioequivalence, EMA, 2010.
11. Directive 2001/83/EC of the European Parliament and of the Council of 6 November 2001 on the Community Code Relating to Medicinal Products for Human Use.
12. Generic Prescribing in Epilepsy. Is it Safe? P. Crawford, W. Hall, B. Chappell et al., Seizure 1996; 5: 1-5.
13. ICH Harmonised Tripartite Guideline: The Common Technical Document for the Registration of Pharmaceuticals for Human Use: Quality M4Q(R1). – Geneva: ICH, 2002.
14. ICH Harmonised Tripartite Guideline: The Common Technical Document for the Registration of Pharmaceuticals for Human Use: Safety. M4S(R2). – Geneva: ICH, 2002.
15. ICH Harmonised Tripartite Guideline: The Common Technical Document for the Registration of Pharmaceuticals for Human Use: Efficacy. M4E(R1). – Geneva: ICH, 2002.
16. Joint Statement between The International Pharmaceutical Federation (FIP) and the International Federation of Pharmaceutical Manufacturers Associations (IFPMA): Ensuring Quality and Safety of Medicinal Products to Protect the Patient. – Barcelona: FIP, IFPMA, 1999.
17. WHO Technical Report Series, No. 902, 2002. Annex 11: Guidance on the Selection of Comparator Pharmaceutical Products for Equivalence Assessment of Interchangeable Multisource (Generic) Products.
18. WHO Technical Report Series, No. 937, 2006. Annex 7: Multisource (Generic) Pharmaceutical Products: Guidelines on Registration Requirements to Establish Interchangeability.
Конечной целью лекарственной политики в любой стране мира является обеспечение населения безопасными, эффективными, качественными и доступными медикаментозными средствами. Один из ключевых моментов в этой политике – широкое использование генерических лекарств.
Ю.С. Рудык, Институт терапии имени Л.Т. Малой АМН Украины, г. Харьков
 Наиболее часто генерики применяются при социально значимых заболеваниях, имеющих высокую распространенность (артериальной гипертонии, хронической сердечной недостаточности, туберкулезе, сахарном диабете и др.). В связи с этим очевидно, что благоприятного влияния на течение и исход социально значимых заболеваний можно добиться только при использовании относительно доступных и высококачественных генериков .
Наиболее часто генерики применяются при социально значимых заболеваниях, имеющих высокую распространенность (артериальной гипертонии, хронической сердечной недостаточности, туберкулезе, сахарном диабете и др.). В связи с этим очевидно, что благоприятного влияния на течение и исход социально значимых заболеваний можно добиться только при использовании относительно доступных и высококачественных генериков .
Согласно определению ВОЗ, под термином «генерик» понимают лекарственный препарат, используемый в медицинской практике взаимозаменяемо с инновационным (оригинальным) средством, производящийся, как правило, без лицензии от компании-создателя и реализуемый после истечения срока действия патента или других исключительных прав.
Генерический препарат должен удовлетворять следующим критериям :
- содержать такой же активный ингредиент, как и оригинальный препарат;
- иметь аналогичную биодоступность;
- выпускаться в той же лекарственной форме;
- сохранять качество, эффективность и безопасность;
- не иметь патентной защиты;
- иметь более низкую стоимость по сравнению с оригинальным препаратом;
- соответствовать фармакопейным требованиям, производиться в условиях GMP (надлежащей производственной практики);
- иметь те же показания к применению и меры предосторожности.
Как показывает клиническая практика, лекарственные препараты, содержащие одинаковые активные ингредиенты в тех же фармацевтических формах и дозах, но производящиеся на различных предприятиях, могут существенно отличаться как по терапевтической эффективности, так и по частоте возникновения побочных реакций, предусмотренных в инструкциях по их медицинскому применению.
В директиве ЕС 2001/83 дается также определение по существу аналогичных препаратов. Лекарственный препарат является по существу аналогичным оригинальному препарату, если он удовлетворяет критериям одного и того же количественного и качественного состава относительно действующих веществ, одной и той же лекарственной формы и является биоэквивалентным, если только с научной точки зрения не очевидно, что он отличается от оригинального препарата относительно безопасности и эффективности.
Одним из главных вопросов как для врача, так и для пациента является проблема взаимозаменяемости генерического и оригинального препаратов .
Международное сообщество, национальные службы здравоохранения заинтересованы в разработке и внедрении в практику научно обоснованных критериев оценки эффективности и безопасности генерических препаратов, которые производятся различными фирмами.
Согласно современным представлениям, соответствие генерика и препарата-бренда основывается на трех важнейших компонентах, обозначаемых как фармацевтическая, фармакокинетическая и терапевтическая эквивалентность .
В странах Европы считается, что лекарственные препараты являются фармацевтически эквивалентными , если они содержат аналогичные активные субстанции в одинаковом количестве и в одинаковой лекарственной форме, отвечают требованиям одних и тех же или сходных стандартов .
По американскому определению, фармацевтически эквивалентные лекарственные препараты содержат одинаковые активные ингредиенты в одинаковой лекарственной форме, предназначены для одного способа введения и идентичны по силе действия или концентрации активных веществ .
Но фармацевтически эквивалентные средства не обязательно будут являться терапевтически эквивалентными, т.е. такими, после применения которых в одинаковой молярной дозе воздействие с точки зрения эффективности и безопасности фактически одинаково. Так, зарегистрированный в Российской Федерации эритромицин при внутривенном введении с высокой частотой вызывал тромботические осложнения, тогда как в странах Европы эритромицин компании Abbott широко используется для внутривенного введения, считаясь при этом наиболее безопасным антибиотиком-макролидом для внутривенных инфузий .
Важную роль в безопасности применения лекарственных средств играют вспомогательные вещества. При создании генерических препаратов следует требовать сохранения оригинального состава вспомогательных вещества, который, однако, не всегда известен. Использование вспомогательных ингредиентов в препаратах-генериках регламентируется на основе рекомендаций ВОЗ .
При оценке фармакокинетической эквивалентности (или биоэквивалентности) сопоставляются особенности всасывания и распределения лекарств в организме человека. Согласно определению ВОЗ, «два лекарственных препарата считаются биоэквивалентными, если они фармацевтически эквивалентны, имеют одинаковую биодоступность и при назначении в одинаковой дозе обеспечивают должную эффективность и безопасность».
Определения, принятые в странах Европейского Союза (ЕС) и в США, несколько отличаются.
Согласно европейской формулировке, два лекарственных препарата биоэквивалентны, если они фармацевтически эквивалентны или альтернативны и если их биодоступность (скорость и степень всасывания) после введения в одинаковой молярной дозе сходна в такой степени, что их эффективность и безопасность в основном одинаковы .
В соответствии с американским определением, биоэквивалентные лекарственные препараты – это фармацевтически эквивалентные или фармацевтически альтернативные средства, которые имеют сравнимую биодоступность при исследовании в сходных экспериментальных условиях .
Исследование биоэквивалентности, по сути, представляет (для перорально применяемых препаратов) испытание сравнительной биодоступности. Для каждого исследуемого лекарства должны быть определены основные фармакокинетические параметры, характеризующие полноту абсорбции: площадь под кривой «концентрация–время» (AUC), скорость абсорбции (C max , T max) и скорость выведения активного вещества (K el , T 1/2). Для заключения об отсутствии различий в этих параметрах применяется дисперсионный анализ и выполняется расчет 90% доверительных интервалов. Для подтверждения эквивалентности требуется, чтобы 90% доверительные интервалы соотношений параметров биодоступности исследуемого препарата не выходили за пределы -80 и +125% показателей референтного препарата.
При этом важно заметить, что нельзя говорить о биоэквивалентности препаратов, если неизвестно наверняка, где и как произведено лекарственное средство. Если нет уверенности в том, что производственный участок, где выпускается этот препарат, соответствует требованиям GMP, бессмысленно заниматься изучением биоэквивалентности, как и другими клиническими испытаниями, потому что качество препаратов не сохраняется от серии к серии. В глобальном смысле GMP – это пошаговое, планомерно-поэтапное «встраивание» качества в препарат. В этом плане исследование биоэквивалентности – лишь часть общей системы обеспечения качества лекарственных средств.
Все генерики должны иметь доказанную биоэквивалентность, поскольку теоретически только биоэквивалентные лекарственные препараты могут обладать сходными клинической эффективностью и профилем безопасности.
В 1984 г. президентом США был подписан закон, требовавший от FDA (Управление по контролю за пищевыми продуктами и лекарственными средствами) сделать общедоступным перечень одобренных рецептурных и безрецептурных лекарственных средств. Этим законом впервые было введено новое допущение, что биоэквивалентные препараты являются терапевтически эквивалентными и поэтому взаимозаменяемыми. Издание «Получившие разрешение на маркетинг лекарственные средства с оценками терапевтической эквивалентности» (Approved Drug Products with Therapeutic Equivalence Evaluations) – список, который принято называть «Оранжевой книгой» (Orange Book) , – идентифицирует препараты, одобренные FDA на основании их безопасности и эффективности. Говоря о статусе «Оранжевой книги», необходимо отметить, что информируя о своей оценке терапевтической эквивалентности препаратов при помощи списка, FDA предлагает свои рекомендации общественности, специалистам и уполномоченным органам по выбору лекарственного средства. Такую оценку не нужно рассматривать как запрет использовать тот или иной препарат либо как свидетельство того, что один их них предпочтительнее другого. «Оранжевая книга» служит большей частью не для различения многоисточниковых лекарственных средств между собой, а информирует о том, удалось ли с помощью доступных инструментов решить проблему доказательства их терапевтической эквивалентности референтному препарату или нет. Терапевтическая эквивалентность является научным суждением, тогда как практика генерической замены, имеющая целью экономию средств, основывается также на социальных и экономических аспектах.
Появление «Оранжевой книги» связано с тем, что в целях экономии средств в системе здравоохранения практически во всех штатах США были приняты законы и/или нормативные акты, стимулирующие выполнение генерической замены. Претворение в жизнь этих законов требовало создания позитивного или негативного перечня лекарств (тех, которыми либо можно, либо нельзя заменять оригинальный препарат). Специалисты FDA создали единый формуляр лекарственных средств, в котором оценка терапевтической эквивалентности препаратов была представлена в виде буквенного кода. Система буквенных кодов, описывающих терапевтическую эквивалентность, позволяет быстро определить, установлена ли биоэквивалентность определенного препарата референтному (первая буква), и получить дополнительную информацию об оценке FDA (вторая буква). Две основные категории, к которым могут быть отнесены генерические препараты, обозначены буквами А и В. К категории А относят препараты, терапевтически эквивалентные другим фармацевтически эквивалентным продуктам, для которых:
- нет известных или предполагаемых проблем с биоэквивалентностью; они обозначаются буквами AA, AN, AO, AP или AT в зависимости от лекарственной формы;
- действительные или потенциальные проблемы с биоэквивалентностью могут быть разрешены путем адекватных доказательств биоэквивалентности; в таких случаях применяют обозначение АВ.
При помощи кода В обозначают препараты, которые FDA в настоящее время считает неэквивалентными терапевтически другим фармацевтически эквивалентным продуктам, то есть действительные или потенциальные проблемы биоэквивалентности не могут быть разрешены путем адекватного установления биоэквивалентности. Часто проблема заключается в определенной лекарственной форме, а не в действующем веществе. В таких случаях применяют обозначения BC, BD, BE, BN, BP, BR, BS, BT, BX или B.
В свое время FDA опубликовало проект руководства по вопросам деятельности фармацевтических компаний, а также предприятий, которые являлись собственностью или подчинялись влиянию распространителей (так называемых спонсоров) медицинской продукции. Необходимость проведения публичных слушаний и обсуждения проекта была обусловлена тем, что отдельные лица и группы людей обращались в государственные законодательные органы, фармацевтические организации, а также в комитеты по контролю за использованием лекарственных средств, выражая озабоченность по поводу проблемы взаимозаменяемости некоторых лекарств, в частности препаратов с ограниченным терапевтическим индексом. Особенно их интересовало, не изменятся ли показатели безопасности и эффективности таких лекарств, если вместо препарата известной фирмы-производителя начать применять препарат, признанный FDA терапевтически эквивалентным, но не защищенный зарегистрированной торговой маркой. С разъяснениями на эту тему в 1998 г. было опубликовано письмо специального уполномоченного по делам здравоохранения FDA Стюарта Л. Найтингейла. Ниже приводится его резюме : «Основываясь на определении терапевтической эквивалентности препаратов, FDA сделало заявление:
- дополнительные клинические тесты не обязательны при замене препарата известной фирмы средством с незарегистрированной торговой маркой;
- не нужно предпринимать специальные меры предосторожности при изменении формулы или процесса производства препарата при условии, что эти изменения одобрены FDA в соответствии с законами и правилами FDA;
- как указано в «Оранжевой книге», по мнению FDA, можно ожидать, что препараты, признанные терапевтически эквивалентными, будут иметь одинаковый клинический эффект, независимо от того, является ли препарат известным или новым;
- нет необходимости рассматривать любой класс лекарственных средств отлично от другого класса, если FDA определила терапевтическую эквивалентность для препаратов, о которых идет речь».
Согласно FDA, терапевтически эквивалентными считаются препараты, удовлетворяющие следующим общим требованиям:
а) доказаны их эффективность и безопасность;
б) они фармацевтически эквивалентны, а именно:
- содержат одинаковое количество идентичных активных ингредиентов в одинаковой лекарственной форме и предназначены для одного способа введения;
- соответствуют требованиям по силе действия, качеству, чистоте и идентичности;
в) являются биоэквивалентными, а именно:
- нет известных или потенциальных проблем с биоэквивалентностью и они соответствуют стандарту in vitro или
- если имеющиеся известные или потенциальные проблемы могут быть устранены проведением исследований биоэквивалентности;
г) адекватные указания в инструкции;
д) произведены в соответствии с требованиями GMP.
Согласно определению ВОЗ, два лекарственных средства считаются терапевтически эквивалентными, если они фармацевтически эквивалентны, имеют одинаковую биодоступность лекарственного вещества и после введения в одинаковой молярной дозе их действие обеспечивает соответствующую эффективность и безопасность .
Таким образом, терапевтическая эквивалентность является основным требованием взаимозаменяемости лекарственных препаратов .
Определение биоэквивалентности лекарственных средств – основной критерий медико-биологического контроля качества генерических препаратов, принятый для стран ЕС , США , Российской Федерации и др.
Считается, что если биоэквивалентность препаратов доказана, нет необходимости проводить дополнительные клинические испытания генерических средств, поскольку наличие биоэквивалентности свидетельствует, что все показатели эффективности и безопасности исследуемого препарата сопоставимы. Испытания с целью установления биоэквивалентности – это клинические исследования, субъектами которых являются здоровые добровольцы или больные, которым показано назначение исследуемого лекарственного средства.
Оценка биоэквивалентности генерических препаратов жестко регламентируется соответствующими международными и национальными стандартами. В настоящее время в Украине в связи с интенсивным расширением фармацевтического рынка возрастает конкуренция среди аналогов лекарственных средств различного производства. Биоэквивалентность многих из них (особенно это касается отечественных лекарственных средств) не доказана. Проведенные клинические испытания по ограниченной программе этих препаратов не всегда могут дать достаточно объективную информацию об их эффективности и безопасности .
В методических рекомендациях ВОЗ по определению взаимозаменяемости аналогичных препаратов, доступных из различных источников (так называемые многоисточниковые лекарственные средства), отмечается, что для подтверждения терапевтической эквивалентности чаще всего используется биоэквивалентность . Вместе с тем возможны и другие подходы , а именно:
- сравнительное определение фармакодинамических характеристик (например, расширение зрачка, изменение сердечного ритма или артериального давления), когда фармакодинамический отклик легче измеряем или более надежен, чем фармакокинетические параметры, или для лекарственных средств местного действия;
- сравнительные клинические испытания в ограниченном объеме, когда ни фармакокинетические, ни фармакодинамические исследования не дают убедительных доказательств;
- испытания in vitro, например определение растворимости дозированной формы (dissolution test), в том числе и в форме профиля растворимости, установленного по нескольким точкам.
Наконец, в ряде случаев специальных доказательств терапевтической эквивалентности не требуется, к примеру при условии, что все химические (например, профиль примесей), фармацевтические (например, стабильность) и производственные (GMP) показатели соответствуют таковым избранного эталона. Иными словами, считается, что в этих случаях соответствие технических параметров само по себе гарантирует терапевтическую эквивалентность. Во всех случаях речь идет о сравнительных испытаниях с препаратами, терапевтическая эффективность которых считается доказанной.
Исходя из вышесказанного очевидно, что терапевтическая эквивалентность включает в себя фармацевтическую эквивалентность и один из критериев :
- изучение биоэквивалентности на людях;
- исследование фармакодинамики на людях;
- клинические испытания;
- тест растворения in vitro (в некоторых случаях).
Производство и контроль качества генериков зависят также от вспомогательных веществ. Требования к ним должны быть такими же, как и к активной субстанции. Любое изменение в составе наполнителей или оболочки лекарства может существенно изменить качество препарата, его биодоступность, привести к токсическим или аллергическим явлениям.
Концепция терапевтической эквивалентности применима лишь к лекарственным средствам, содержащим одинаковые активные ингредиенты, и не распространяется на разные терапевтические препараты, используемые в одинаковых клинических ситуациях (например, парацетамол и ацетилсалициловая кислота, назначаемые при головной боли).
Лекарственное средство, соответствующее вышеуказанным критериям терапевтической эквивалентности, считается таковым, даже если оно отличается по некоторым характеристикам, таким как форма, риска на таблетке, упаковка, эксцепиенты (включая красители, консерванты), срок годности и минимальные отличия в инструкции (например, наличие специфической информации о фармакокинетике), а также условия хранения. Если подобные отличия важны в лечении конкретного пациента, врач может требовать, чтобы ему был отпущен из аптеки конкретный бренд. Не считая этого ограничения, FDA полагает, что препараты, классифицированные как терапевтически эквивалентные, можно заменять, полностью полагаясь на то, что в результате замены сохранятся эффекты и профиль безопасности, ожидаемые от назначенного препарата.
Надо признать, что как в странах ЕС, так и в США многие специалисты ставят под сомнение фармакокинетическую эквивалентность как единственный способ оценки взаимозаменяемости лекарств. В ряде публикаций указывается на существенные методологические недостатки изучения биоэквивалентности лекарственных средств, которые могут привести к тому, что существующие различия между брендовыми и генерическими препаратами не будут выявлены . Согласно европейским требованиям и регламенту FDA, отдельные показатели фармакокинетики могут отличаться до 20%. Считается, что колебания концентрации активного компонента в плазме крови в пределах от -20 до +25% клинически не значимы, однако для пациентов пожилого возраста или других уязвимых групп больных даже такие незначительные изменения концентрации лекарственного вещества могут повышать риск побочных эффектов.
Предполагается, например, что определенные ограничения могут быть связаны с существованием лекарственных средств, характеризующихся сравнительно небольшим разбросом терапевтических концентраций препарата в плазме крови (некоторые антидепрессанты – пароксетин, флуоксетин, циталопрам) и/или нелинейной фармакокинетикой (нормотимики и противоэпилептические препараты) .
В данной ситуации даже небольшие изменения этого параметра, вполне укладывающиеся в допустимые пределы теста биоэквивалентности (от -20 до +25%), могут оказаться значимыми для клинической эффективности и/или переносимости .
Следовательно, возможны значительные расхождения свойств брендовых и генерических лекарственных средств . Например, при показателях биоэквивалентности ниже 100% препарат может оказаться неэффективным. Напротив, при повышении рассматриваемого показателя следует ожидать возрастания числа побочных эффектов. Особые опасения вызывают лекарственные средства с низким терапевтическим индексом (разницей между минимальной эффективной дозой препарата и его максимально токсичной дозой) – дигоксин, фенитоин, карбамазепин, циклоспорин, варфарин. Такая ситуация требует ужесточения и расширения требований к фармакокинетическим исследованиям. Обсуждается вопрос об уменьшении различий в параметрах до 10-15%, что позволит уменьшить число препаратов с пограничными фармакокинетическими параметрами.
Еще одно ограничение накладывает на использование результатов теста биоэквивалентности существование лекарственных средств (сертралин, флуоксетин, хлорпромазин, клозапин) со значительной вариабельностью фармакокинетических показателей, которая зависит, в частности, от сложности процессов метаболизма препарата (система цитохромов, наличие нескольких путей выведения и т.д.). Такая изменчивость может носить «интраиндивидуальный» характер. В одном случае она связана, например, с генетическим полиморфизмом цитохромов, который наблюдается в различных популяциях населения, в другом – с функциональным состоянием этих ферментов, меняющимся у одного и того же человека под влиянием различных внешних факторов (например, употребление грейпфрутового сока). Следовательно, результаты теста биоэквивалентности, выполненного на небольшой группе добровольцев, употреблявших сходную диету, могут оказаться невалидными для реальных клинических условий .
Критически воспринимается и тенденция использовать во время исследования биоэквивалентности единственную суточную дозу препаратов .
Известно, что многие препараты (амиодарон, препараты дигиталиса, психотропные средства) назначаются многократно в течение определенного промежутка времени и для получения клинического эффекта требуется достижение устойчивой (терапевтической) концентрации препарата в плазме крови и/или ткани, которая может быть значительно выше той, что используется при проведении исследований биоэквивалентности на здоровых добровольцах .
Следует также иметь в виду, что в реальной клинической практике воспроизведенные лекарственные средства длительное время принимают пациенты разного возраста, пола, массы тела, часто страдающие коморбидной (сопутствующей) патологией. В такой ситуации фармакокинетические свойства брендовых и генерических лекарственных средств в силу существования между ними даже небольших химических отличий могут существенно различаться. Определенное значение приобретает патология желудочно-кишечного тракта. У пациентов с таким заболеванием достаточно сложный механизм всасывания лекарственного вещества легко нарушается. При этом даже незначительные различия в химическом составе брендового и генерического препаратов могут привести к нарушению их биоэквивалентности .
В частности, может возникнуть ситуация, когда инертные составы (наполнители), используемые в генериках, при назначении в одной-единственной дозе, не влияя на всасывание, распределение и метаболизм препаратов, при длительном применении могут оказывать воздействие на функциональное состояние желудочно-кишечного тракта, печени или почек таким образом, что фармакокинетическая эквивалентность препаратов существенно нарушается .
Как пример можно привести различные составы вспомогательных веществ оригинальных и генерических препаратов ницерголина, широко применяемых пациентами разного возраста, в том числе пожилыми больными, часто страдающими широким спектром сопутствующих заболеваний внутренних органов.
С наличием сопутствующей соматической патологии связана и другая проблема, значительно затрудняющая клиническое использование результатов теста биоэквивалентности. В отличие от здоровых добровольцев пациенты с сопутствующей патологией часто вынуждены принимать различные соматотропные лекарственные средства, в частности усиливающие или ослабляющие перистальтику, влияющие на разрушение лекарственного вещества в кишечнике. Не исключено, что это влияние в силу существующих, хотя и минимальных, отличий химического состава оригинального и генерического препаратов может оказаться неоднозначным. Соответственно возникают условия для изменения биоэквивалентности этих препаратов .
Рассмотренные возражения не являются лишь теоретическими соображениями. В соответствующих публикациях имеется много сведений о результатах перепроверки биоэквивалентности различных препаратов. Эти данные свидетельствуют о том, что значительная часть генериков не выдерживает такого тестирования. Так, проведенный в Великобритании в 1995-1996 гг. анализ 2427 воспроизведенных лекарственных средств обнаружил 228 существенных различий . Не менее поразительные данные получены в США. FDA выявило, что до 20% имеющихся на территории страны брендовых и генерических препаратов не являются биоэквивалентными и, следовательно, не могут быть взаимозаменяемыми .
Приводятся примеры клинической неэквивалентности препаратов эналаприла. Показано, что клиническая эффективность по достижению целевого уровня артериального давления у пациентов с артериальной гипертензией 4 генерических эналаприлов известных производителей была ниже, чем у оригинального препарата (Ренитек, MSD). Исследуемые генерики были фармакокинетически эквивалентны Ренитеку. На основании полученных результатов авторы сделали вывод о неодинаковой терапевтической эквивалентности воспроизведенных препаратов эналаприла .
О терапевтической неэквивалентности оригинального индапамида (Арифон, Servier) и его генериков у пациентов с артериальной гипертензией сообщают В.И. Петров и соавт. , при этом фармакокинетические профили сравниваемых препаратов совпадали .
Особое значение эквивалентность генериков имеет для антимикробных препаратов, так как низкая антимикробная активность может приводить к снижению клинической эффективности терапии, что особенно важно при лечении тяжелых больных, и быстрому распространению резистентных форм микробов. Недавно проведенное исследование микологической активности оригинального флуконазола (Дифлюкан, Pfizer) и генерических препаратов показало, что активность генериков против различных видов грибов рода Candida в 2 раза ниже, чем у Дифлюкана. При этом генерики были биоэквивалентны оригинальному препарату .
В одной из публикаций приводятся данные сравнительного анализа качества оригинального кларитромицина производства компании Abbott и 40 его генериков из 13 стран Азии и Латинской Америки. Оказалось, что в 8 препаратах содержание активного вещества не соответствовало стандартам компании-разработчика, у 28 генериков количество высвобождающегося при растворении активного компонента было значительно ниже, чем у оригинального, хотя все они имели соответствующую спецификацию. У 24 из 40 препаратов был превышен рекомендованный компанией Abbott 3% лимит посторонних примесей.
В 10 раз увеличено количество твердых частиц в 4 воспроизведенных препаратах цефотаксима по сравнению с оригинальным лекарственным средством (Клафоран, Hoechst). Указанные частицы, содержащиеся в генериках, могут нарушать микроциркуляцию в ишемизированных тканях и способствовать развитию респираторного дистресс-синдрома и полиорганной недостаточности у тяжелых пациентов .
В литературе представлены результаты сравнения брендового и генерического клозапина (Клозарил, Novartis Pharmaceuticals и клозапин, Zenith Goldline Pharmeceuticals). В ходе исследования было установлено, что расхождение между указанными психотропными препаратами по фармакокинетическим параметрам наблюдается у 40% пациентов, страдающих шизофренией .
Выявлены существенные различия в биоэквивалентности между брендовыми препаратами амитриптилина гидрохлорида, нортриптилина гидрохлорида, дезипрамина, тримипрамина малеата и их генериками .
Проведено более 100 исследований, посвященных биоэквивалентности различных генериков фенитоина, препаратов вальпроевой кислоты, в которых обнаружены существенные расхождения по фармакокинетическим параметрам оригинальных и воспроизведенных лекарственных средств .
Говоря о терапевтической эквивалентности, следует упомянуть исследование R. Mofsen и соавт., в котором описываются 7 случаев неудачной замены у пациентов со стабильным психическим состоянием, находившихся в психоневрологическом интернате, брендового клозапина на его генерик. Подчеркивается, что указанное изменение терапии было неожиданно произведено аптекой и ни врачи, ни медицинский персонал учреждения не знали об этом. Для них оказалось полной неожиданностью возобновление у пациентов психотических расстройств, выраженность которых в 5 из 7 случаев потребовала неотложных мер по переводу больных в психиатрический стационар. Сообщается об аналогичном случае при переводе с брендового пароксетина (паксила) на его генерик .
В проведенном недавно опросе неврологов (301 опрошенный), работающих в США, установлено, что при переводе с брендовых противоэпилептических препаратов на генерические 204 (67,8%) из них наблюдали возобновление судорожных припадков, 168 (55,8%) отмечали усиление побочных эффектов .
Описаны 11 наблюдений, в которых после замены брендового ламотриджина на его генерики контроль над эпилептическими припадками был утрачен.
В результате этих исследований в ряде стран, в том числе в Норвегии, приняты решения, ограничивающие перевод пациентов с брендовых противоэпилептических на генерические препараты, а в Германии эта процедура вообще не рекомендуется .
В ряде контролируемых исследований показано, что при переходе с брендового карбамазепина на его генерик отмечается внезапное возобновление судорожных припадков .
В другой работе, опубликованной в Американском кардиологическом журнале в мае 2000 г., приводится мнение 64 экспертов-электрофизиологов, членов Североамериканского общества стимуляции электрофизиологии, которые сообщают о 32 случаях развития рецидива аритмии (фибрилляции желудочков, желудочковой тахикардии, фибрилляции предсердий и предсердной тахикардии) при замене брендового антиаритмического препарата амиодарона (Кордарон, Sanofi-Synthelabo) на его генерики.
Следует отметить, что имеются также и публикации о терапевтической эквивалентности оригинальных и генерических лекарственных средств. В одном из рандомизированных двойных слепых исследований изучали две параллельные группы амбулаторных пациентов с хронической шизофренией, получавших брендовый флуфеназина деканоат. Первая группа была переведена на его генерик, вторая – оставлена на оригинальном препарате. Через 12 недель в обеих группах отсутствовала какая-либо значимая динамика в состоянии, определяемая по специальной шкале позитивного и негативного синдрома .
Говоря о хронических заболеваниях, необходимо заметить, что многие из них имеют тенденцию к рецидивированию. Ввиду этого современные рекомендации предусматривают наряду с купирующей осуществлять и длительную поддерживающую терапию. На практике нередко наблюдается ситуация, когда купирующая терапия, осуществляемая чаще всего в стационаре, проводится оригинальным лекарственным средством. В дальнейшем после выписки пациента этот препарат в силу «экономических» соображений нередко заменяется на его генерик. В свете представленных выше данных очевидно, что рассматриваемая замена возможна лишь при наличии уверенности в фармацевтической, фармакокинетической и терапевтической эквивалентности оригинального и воспроизведенного лекарственных средств.
Имеются сообщения о том, что появление на фармацевтическом рынке воспроизведенных лекарственных средств не всегда ведет к снижению прямых затрат здравоохранения. В недавно проведенном канадском исследовании проанализировано, что 11% разница в количестве рецидивов, наблюдающаяся при лечении больных генерическим и оригинальным клозапином, сводит на нет преимущество генерика в цене. Аналогичные данные были получены и для противоэпилептических препаратов .
Приведенные данные, как и многие другие, по мнению главного клинического фармаколога МЗ РФ профессора Ю.Б. Белоусова, развеивают миф о дешевизне генериков, так как затраты при их применении гораздо выше, чем при использовании оригинальных препаратов. Вопреки расхожим утверждениям о том, что воспроизведенные лекарственные средства снижают прямые затраты на лечение, способствуют развитию конкурентной борьбы и снижению цен на препараты-бренды и даже являются одним из способов внедрения экономически эффективных медицинских технологий в клиническую практику, некоторые современные исследования свидетельствуют об обратном.
Ученый полагает, что переход с недорогих генериков на оригинальные препараты выгоден как для пациентов, так и для общества в целом. Он считает, что недопустимо переносить данные по эффективности и безопасности, полученные на оригинальных препаратах, на их копии. Лишь наличие полной информации о соблюдении требований GMP при производстве генерика, его фармакокинетическая и терапевтическая эквивалентность при сравнении с оригинальным препаратом делают обоснованным поиск фармакоэкономических преимуществ генерика. В противном случае формально выгодные ценовые показатели могут обернуться огромными дополнительными расходами, например на лечение нежелательных побочных явлений. По мнению Ю.Б. Белоусова, практика, сложившаяся в РФ, разрешающая медицинское применение генерика на основании данных только его биоэквивалентности, неверна. Для определения терапевтической эквивалентности необходимо проведение как ограниченных, так и крупных клинических исследований эффективности генерика при конкретном заболевании, изучение сравнительной эффективности оригинального и воспроизведенного препаратов с использованием четких конечных критериев. Терапевтическая эквивалентность означает и организацию исследований профиля безопасности генериков с интенсивным мониторингом в течение 5 лет после регистрации нежелательных эффектов.
Очевидно, что оригинальные препараты всегда будут противопоставляться генерическим, но их конкуренция на фармацевтическом рынке должна базироваться на строгом соблюдении требований к качеству производства как оригинальных, так и воспроизведенных лекарственных средств, на результатах анализов биоэквивалентности, а также данных клинических исследований. Поэтому широкое использование генерических препаратов в клинической практике должно основываться на доступных практикующим врачам ясных указаниях на их фармацевтическую, фармакокинетическую и в первую очередь терапевтическую эквивалентность оригинальным препаратам.
Список литературы находится в редакции
Терапевтическая эквивалентность воспроизведенного препарата (дженерика) и как ее доказать.
Н.П.Кутишенко1, С.Ю.Марцевич1,2, И.В.Вашурина1
1ФГУ ГНИЦ ПМ Минздравсоцразвития России, Москва
2Кафедра доказательной медицины Первого МГМУ им. И.М.Сеченова
Проблема эффективности и безопасности препаратов-копий (воспроизведенных препаратов, дженериков) продолжает беспокоить ученых, врачей, общественность. К ней постоянно обращаются на научных конференциях и симпозиумах, в средствах массовой информации , ей посвящаются специальные научные исследования, в которых иногда участвуют тысячи больных, например, исследование ОРИГИНАЛ (Оценка эффективности пеРевода с Индапамидов ГенерИков На Арифон ретард у пациентов с АртериаЛьной гипертензией) . И все это несмотря на то, что научная часть этой проблемы в основном уже давно решена в многочисленных исследованиях, а ее практическая часть нашла отражение в целом ряде нормативных документов, о которых речь пойдет ниже. Характерно, что в зарубежной научной литературе сейчас крайне редко появляются публикации, посвященные сравнительной оценке оригинальных препаратов и дженериков, хотя совсем недавно таких публикаций было значительно больше .
Безусловно, определенные неясности в отношении эффективности и безопасности некоторых дженериков остаются, однако они, по нашему мнению, отражают в первую очередь проблемы с соблюдением тех необходимых условий, которые, по современным представлениям, обеспечивают терапевтическую эквивалентность препарата-дженерика.
Цель данной публикации как раз и заключается в том, чтобы напомнить об основных принципах оценки терапевтической эквивалентности воспроизведенных лекарственных препаратов.
Что такое дженерик (воспроизведенный препарат)
Как это ни покажется странным, но единого определения понятия «дженерик» до сих пор не существует: ВОЗ (Всемирная Организация Здравоохранения), FDA (Food and Drug Administration), EMEA (European Medicines Agency), министерства здравоохранения различных стран предлагают свои определения для воспроизведенного препарата, а также критерии, на основании которых дженерик можно считать терапевтически эквивалентным оригинальному препарату. В целом эти критерии совпадают, однако есть определенные различия в оценке значимости и необходимости проведения исследований по терапевтической эквивалентности для доказательства соответствия дженерика оригинальному препарату как по эффективности, так и по безопасности.
Без сомнения, наиболее четкая, продуманная и научно обоснованная система оценки эквивалентности дженериков на сегодняшний день существует в США, которая отражена в документах FDA. В соответствии с определением FDA, терапевтическая эквивалентность устанавливается в ходе исследований фармацевтической эквивалентности и биоэквивалентности. Если сомнений в эквивалентности нет, то препарату присваивается соответствующий код, начинающийся с буквы «А», что также означает, что он может рассматриваться как возможный референсный препарат (т.е. препарат сравнения). Если данные биоэквивалентности не исключают потенциальных сомнений в отношении терапевтической эквивалентности фармацевтически эквивалентных препаратов или изучение биоэквивалентности не проводилось (н-р, для препаратов местного действия), то код оценки терапевтической эквивалентности начинается с буквы «В». Большинство дженериков в соответствии с данной системой кодировки, как правило, получают код «АВ» - это означает, что различия между препаратами потенциально возможны, но эквивалентность подтверждается результатами адекватно выполненных in vitro или/и in vivo исследований. Следует отметить, что проведение специальных клинических исследований, подтверждающих терапевтическую эквивалентность оригинального препарата и дженерика, не предполагается .
ВОЗ определяет терапевтическую эквивалентность оригинального препарата и дженерика (мультиисточникового фармацевтического продукта) несколько иначе. В соответствии с требованиями ВОЗ два фармацевтических продукта считаются терапевтически эквивалентными в том случае, если они фармацевтически эквивалентны (или являются фармацевтически альтернативными) и после введения в одинаковой молярной дозе их эффект в отношении эффективности и безопасности совершенно одинаков при одинаковом способе назначения и при одинаковых показаниях. Это должно быть продемонстрировано с помощью соответствующих исследований биоэквивалентности, таких как фармакокинетические, фармакодинамические, клинические или в исследованиях in vitro .
С точки зрения EMEA (European Medicines Agency) исследования по биоэквивалентности необходимы не только для того, чтобы продемонстрировать сходство между дженериком и оригинальным препаратом по основным фармакокинетическим показателям. Такие исследования предоставляют реальную возможность переноса данных об эффективности и безопасности, полученных для оригинального препарата, на дженерик, при этом проведение исследований терапевтической эквивалентности не предполагается (исключение - биологические лекарственные препараты) .
Российский Федеральный закон «Об обращении лекарственных средств» вводит понятие воспроизведенного лекарственного средства, однако входит в некоторое противоречие с документами других стран. В соответствии с Федеральным законом Российской Федерации от 12 апреля 2010 г. N 61-ФЗ » при проведении процедуры экспертизы воспроизведенных лекарственных средств (к ним как раз и относятся дженерики) должна быть представлена информация, полученная при проведении клинических исследований лекарственных препаратов и опубликованная в специализированных печатных изданиях, а также документы, содержащие результаты исследования биоэквивалентности и (или) терапевтической эквивалентности. Если говорить об исследованиях терапевтической эквивалентности лекарственных препаратов, то под этим термином понимается вид клинического исследования, проведение которого осуществляется для выявления одинаковых свойств лекарственных препаратов определенной лекарственной формы, а также наличия одинаковых показателей безопасности и эффективности лекарственных препаратов, одинаковых клинических эффектов при их применении .
Относительно вопроса о подтверждении терапевтической эквивалентности сложились определенные противоречия с правилами FDA, также отсутствуют документы, определяющие порядок проведения и критерии оценки результатов таких клинических испытаний. Если обратиться к проверенным временем правилам FDA по определению терапевтической эквивалентности, то для этого должны быть в обязательном порядке соблюдены пять условий: 1) препараты должны быть признаны эффективными и безопасными, 2) они должны быть фармацевтически эквивалентными, включая соответствие по количеству активных ингредиентов, их чистоте, качеству, идентичности, 3) соответствовать стандартам биоэквивалентностии при участии в исследовании не менее 24-36 добровольцев, 4) правильно промаркированы и, что не менее важно, 5) произведены в соответствии с требованиями GMP (Good Manufacturing Practice) .
Значимость исследований по терапевтической эквивалентности
Однако, несмотря важность показателей биоэквивалнтности при регистрации дженерика, результаты клинических исследований для доказательства эквивалентности сохраняют определенную значимость. В большей мере это касается аналогов фармацевтических средств биологического происхождения (т.н. биосимиляров или биодженериков). Для них исследования терапевтической эквивалентности - одно из условий регистрации. В скором времени такие препараты все чаще будут появляться на фармацевтическом рынке, поскольку сроки действия патентов на ряд оригинальных биопрепаратов (в т.ч. на низкомолекулярные гепараины) заканчиваются. В связи с этим некоторые дженериковые компании приступили к разработке производства биосимиляров, несмотря на то, что химическая структура и технология получения биосимиляров значительно сложнее, чем традиционных химических лекарственных средств. Поскольку биосимиляры имеют сложную трехмерную пространственную структуру, их количественное содержание в биологических жидкостях точно охарактеризовать достаточно трудно, поэтому принято считать, что для таких препаратов обычных исследований биоэквивалентности явно недостаточно. Это заставляет регуляторные органы требовать от производителей биосимиляров проведения как доклинических (токсикологических, фармакокинетических и фармакодинамических), так и клинических исследований (полного представления данных по эффективности и безопасности препарата), а также данных по изучению иммуногенности . К препаратам биологического происхождения относятся гормоны, цитокины, факторы свертывания крови, моноклональные антитела, ферменты, вакцины и препараты, созданные на базе клеток и тканей и т.д.
«Дженериковая замена»
Необходимо отметить, что различия в лечебном эффекте оригинальных лекарств и дженериков или различных дженериков между собой, в принципе, допускаются рядом международных документов. Достаточно давно был введен термин «дженериковая замена», под которой понимают отпуск лекарственного препарата, коммерческое название которого отличается от выписанного врачом, а химический состав и дозировка действующего начала - идентична. В документах Всемирной Медицинской Ассамблеи делается предупреждение, что при отпуске препаратов, не полностью идентичных по химическому составу, биологическому действию или терапевтической эффективности пациент может столкнуться с неадекватным эффектом, т.е. с побочными реакциями или с недостаточной лечебной эффективностью. В этом документе обращается особое внимание на то, что государственные службы контроля должны информировать врачей о степени химической, биологической и терапевтической идентичности препаратов, выпускаемых одним или разными производителями, а службы контроля качества, существующие на предприятиях-производителях лекарств, обязаны следить за неуклонным соответствием выпускаемых препаратов стандартам химических и биологических свойств .
Возникает вопрос, почему, несмотря на установившиеся методы контроля дженериков, на рынок нередко попадают такие из них, которые явно не полностью соответствуют оригинальным препаратам ни по эффективности, ни по безопасности, а иногда и по тому и другому показателю . Такая ситуация, к сожалению, достаточно типична для нашей страны . Окончательного ответа на этот вопрос пока нет, но, думается, главное заключается в нарушении тех самых принципов доклинической оценки дженериков, о которых было сказано выше. Хорошо известно, что в России стандарт GMP до сих пор не соблюдается при производстве большей части препаратов, выпускаемых в нашей стране (считается, что переход всех российских производителей лекарств на стандарт качества GMP должен произойти только к январю 2014 года), и уже одно это создает весомую причину для получения несоответствующих по качеству дженериков.
На что должен ориентироваться практический врач при выборе воспроизведенных препаратов
Возникает и более простой вопрос: что делать практическим врачам, выбирая лекарственный препарат, особенно в тех случаях, когда эта терапия является длительной и от качества которой может зависеть судьба больного, например, при вторичной профилактике сердечно-сосудиситых осложнений у кардиологических больных высокого риска. С одной стороны, все нормативные документы, а также экономическая целесообразность заставляют врача воспользоваться в первую очередь именно дженериком (если он зарегистрирован). С другой стороны, ряд четко спланированных клинических исследований (неконтролируемые исследования не в счет) свидетельствует о том, что далеко не все дженерики являются полноценными копиями. Этими фактами умело пользуются фармацевтические компании, утверждающие, что все дженерики - препараты неполноценные и, используя их, врач заведомо идет на назначение менее эффективной терапии .
Большинство российских специалистов, признавая изложенные выше факты, делают вывод о необходимости проведения прямых сравнительных исследований по изучению терапевтической эквивалентности с теми дженериками, которые уже зарегистрированы и чаще всего назначаются в клинике.Отделом профилактической фармакологии ФГУ ГНИЦ ПМ предпринята попытка создания реестра клинических контролируемых рандомизированных исследований, выполненных с дженериками в России .
Таким образом, с одной стороны, нет никаких оснований сомневаться в том, что создание дженерика - полной копии оригинального препарата - дело абсолютно возможное. Однако те или иные отклонения при разработке и производстве дженерика могут отразиться на его качестве. В идеале эти отклонения должны фиксироваться всей системой доклинического контроля, однако на практике, по-видимому, не всегда эта система четко соблюдается, что и приводит к появлению не полностью эквивалентных дженериков. В таких случаях единственным способом подтвердить качество дженерика является проведение методически грамотно спланированных сравнительных клинических испытаний по изучению терапевтической эквивалентности. Результаты таких исследований позволят также более точно ответить на вопрос о рациональности вмешательства как с точки зрения экономической эффективности, так и его доступности.
Список литературы
- Марцевич С.Ю. Копии лекарств, как и копии в искусстве, бывают разными. АИФ. Здоровье 2010; 4: 2-3.
- Марцевич С.Ю. Таблетки на замену. Чем отличаются дешевые лекарства от дорогих. АИФ. Здоровье 2011, 8: 35.
- Карпов Ю.А., Недогода С.В., Кисляк О.А., Деев А.Д. и др. Основные результаты программы ОРИГИНАЛ. Кардиология 2011; 3: 36-41.
- Johnston A, Staylas P, Stergiou G. Effectiveness, safety and cost of drug substitution in hypertension. Br J Clin Pharmacol 70: 3; 320-334.
- www.Guideline on similar biological medicinal products (CHMP/437/04
- World Medical Association Statement on Generic Drug Substitution Adopted by the 41st World Medical Assembly Hong Kong, September 1989 and rescinded at the WMA General Assembly, Santiago 2005
- Рекомендации ВНОК «Рациональная фармакотерапия больных с сердечно-сосудистыми заболеваниями». В составе рабочей группы: Марцевич С.Ю., Аничков Д.А., Белолипецкая В.Г., Концевая А.В., Кутишенко Н.П., Лукина Ю.В., Толпыгина С.Н., Шилова Е.В., Якусевич В.В. Кардиоваск тер профил 2009; 6: приложение 4: 56 c.
- Якусевич В.В. Качественное лекарственное средство: каким оно должно быть. Рациональная фармакотерапия в кардиологии 2006; 4: 41-46.
- Ревельский И.А. Способ сравнительной физиологической оценки фармацевтических субстанций и препаратов на их основе. Вестник Росздравнадзора 2009; 4: 48-51.
- Марцевич С.Ю., Кутишенко Н.П., Деев А.Д. Оригинальные препараты и дженерики в кардиологии. Можно ли решить проблему взаимозаменяемости. Вестник Росздравнадзора 2009; 4: 48-51.