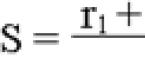Генетические патологии – это редкие врожденные заболевания, которые трудно предусмотреть заранее. Они возникают еще в тот момент, когда происходит формирование эмбриона. Чаще всего они передаются от родителей, но так происходит не всегда. В некоторых случаях нарушения в генах возникают самостоятельно. Одной из таких патологий считается болезнь Брутона. Она относится к первичных иммунодефицитных состояний. Данное заболевание открыто недавно, в середине 20 века. Поэтому оно не полностью изучено докторами. Встречается оно довольно редко, только у мальчиков.
Болезнь Брутона: история изучения
Эта патология относится к Х-сцепленным хромосомных аномалий, что передается на генетическом уровне. Болезнь Брутона характеризуется нарушениями в гуморальном иммунитете. Основным симптомом является восприимчивость к инфекционным процессам. Первое упоминание об этой патологии приходится на 1952 год. В то время американский ученый Брутон изучал анамнез ребенка, что болеет больше 10 раз в 4-летнем возрасте. Среди инфекционных процессов у этого мальчика встречались сепсис, пневмония, менингит, воспаление верхних дыхательных путей. При обследовании ребенка было установлено, что антитела к этим заболеваниям отсутствуют. Другими словами, после перенесенных инфекций иммунного ответа не наблюдалось. Позже, в конце 20 века, болезнь Брутона снова изучалась докторами. В 1993 году врачам удалось определить дефектный ген, который вызывает нарушение иммунитета.
Причины возникновения болезни Брутона
Агаммаглобулинемия (болезнь Брутона) зачастую носит наследственный характер. Дефект считается рецессивным признаком, поэтому вероятность рождения ребенка с патологией составляет 25 %. Носителями мутантного гена являются женщины. Это связано с тем, что дефект локализуется в Х-хромосоме. Тем не менее заболевание передается только мужского пола. Основной причиной агаммаглобулинемии является дефектный белок, что входит в состав гена, кодирующего тирозинкиназу. Кроме этого болезнь Брутона может быть и идиопатической. Это означает, что причина ее появления остается невыясненной. Среди факторов риска, влияющих на генетический код ребенка, выделяют:
патогенез заболевания?
Механизм развития болезни связан с дефектным белком. В норме ген, отвечающий за кодирование тирозинкиназы, участвует в процессе образования В-лимфоцитов. Они представляют собой иммунные клетки, которые отвечают за гуморальную защиту организма. Из-за несостоятельности тирозинкиназы В-лимфоциты не созревают полностью. Вследствие этого они не способны вырабатывать иммуноглобулины – антитела. Патогенез болезни Брутона заключается в полном блокировании гуморальной защиты. В результате этого при попадании инфекционных агентов в организм антитела к ним не производятся. Особенностью этого заболевания является то, что иммунитет способен бороться с вирусами, несмотря на отсутствие В-лимфоцитов. Характер нарушения гуморальной защиты зависит от степени выраженности дефекта.
Болезнь Брутона: симптомы патологии
Патология впервые дает о себе знать в грудном возрасте. Чаще всего болезнь проявляется до 3-4-го месяца жизни. Это объясняется тем, что в данном возрасте организм ребенка перестают защищать материнские антитела. Первыми признаками патологии могут стать болезненная реакция после вакцинации, кожные высыпания, инфекции верхних и нижних дыхательных путей. Тем не менее грудное вскармливание защищает малыша от воспалительных процессов, так как материнское молоко имеет в своем составе иммуноглобулины. Болезнь Брутона манифестирует примерно до 4 лет. В это время ребенок начинает контактировать с другими детьми, посещает детский сад. Среди инфекционных поражений преобладает менинго-, стрепто - и стафилококковая микрофлора. Вследствие этого дети могут быть подвергнуты гнойных воспалений. К наиболее часто встречающимся заболеваниям относят воспаление легких, синусит, отит, гайморит, менингит, конъюнктивит. При несвоевременном лечении все эти процессы могут перейти в сепсис. Также проявлением болезни Брутона могут быть дерматологические патологии. Из-за сниженного иммунного ответа микроорганизмы быстро размножаются на месте ран и царапин. Кроме этого к проявлениям заболевания относятся бронхоэктазы – патологические изменения в легких. Симптомами являются одышка, боли в области груди, иногда кровохарканье. Также возможно появление воспалительных очагов в органах пищеварения, мочеполовой системы, на слизистых оболочках. Периодически наблюдаются припухлость и болезненность в суставах.
Диагностические критерии заболевания
До первого диагностического критерия следует отнести частую заболеваемость. Дети, которые страдают патологией Брутона, переносят более 10 инфекций в год, а также несколько раз в течение месяца. Заболевания могут повторяться или сменять друг друга (отит, тонзиллит, пневмония). При осмотре зева отмечается отсутствие гипертрофии миндалин. То же касается и пальпации периферических лимфатических узлов. Также следует обратить внимание на реакцию малыша после вакцинации. Существенные изменения наблюдаются в лабораторных анализах. В ОАК отмечаются признаки воспалительной реакции (повышенное количество лейкоцитов, ускоренная СОЭ). При этом количество иммунных клеток снижен. Это отражается в лейкоцитарной формуле: малое число лимфоцитов и повышенное содержание нейтрофилов. Важным исследованием является иммунограмма. В ней отражается снижение или отсутствие антител. Этот признак позволяет поставить диагноз. Если у доктора есть сомнения, можно провести генетическое обследование.
Отличия болезни Брутона от схожих патологий
Данную патологию дифференцируют с другими первичными и вторичными иммунодефицитами. Среди них - агаммаглобулинемия швейцарского типа, синдром Ди Джорджи, ВИЧ. В отличие от этих патологий для болезни Брутона характерно нарушение только гуморального иммунитета. Это проявляется тем, что организм способен бороться с вирусными агентами. Данный фактор отличается от агаммаглобулинемии швейцарского типа, при которой нарушается как гуморальный, так и клеточный иммунный ответ. Чтобы провести дифференциальную диагностику с синдромом Ди Джорджи, необходимо сделать рентген грудной клетки (аплазия тимуса) и определить содержание кальция. Для исключения ВИЧ-инфекции проводится пальпация лимфатических узлов, ИФА.
Методы лечения агаммаглобулинемии
К сожалению, полностью победить болезнь Брутона невозможно. К методам лечения агаммаглобулинемии относят заместительную и симптоматическую терапию. Главной целью считается достижение нормального уровня иммуноглобулинов в крови. Количество антител должно быть приближено до 3 г/л. Для этого используют гамма-глобулин из расчета 400 мг/кг массы тела. Концентрация антител должна быть увеличена в период острых инфекционных заболеваний, так как организм не может справиться с ними самостоятельно. Кроме этого, проводится симптоматическое лечение. Чаще всего назначают антибактериальные препараты «Цефтриаксон», «Пенициллин», «Ципрофлоксацин». При кожных проявлениях необходимо местное лечение. Также рекомендуется промывание слизистых оболочек антисептическими растворами (орошение горла и носа).
Прогноз при агаммаглобулинемии Брутона
Несмотря на пожизненную заместительную терапию, прогноз при агаммаглобулинемии благоприятный. Постоянное лечение и профилактика инфекционных процессов сводят к минимуму заболеваемость. Обычно пациенты остаются трудоспособными и активными. При неправильном подходе к лечению возможно развитие осложнений вплоть до сепсиса. В случае запущенных инфекций прогноз неблагоприятный.
Профилактика болезни Брутона
При наличии патологии у родственников или подозрении на нее необходимо провести генетическое обследование еще во время первого триместра беременности. Также к профилактическим мерам следует отнести пребывание на воздухе, отсутствие хронических инфекций и вредного влияния. Во время беременности противопоказаны стрессы. К вторичной профилактике относятся витаминотерапия, введение гамма-глобулина, здоровый образ жизни. Также важно избегать контактов с инфицированными людьми.
Болезнь Брутона , или агаммаглобулинемия Брутона , является наследственным иммунодефицитом, который вызывается мутациями в гене кодирующем тирозинкиназы Брутона. Впервые болезнь была описана Брутоном в 1952 году, в честь которого и был назван дефектный ген. Тирозинкиназы Брутона имеют решающее значение в созревании пре-В-клеток к дифференциации зрелых В-клеток. Ген тирозинкиназы Брутона был обнаружен на длинном плече Х-хромосомы в полосе от Xq21.3 к Xq22, он состоит из 37.5 килобаз с 19 экзонами, которые кодируют 659 аминокислот, именно эти аминокислоты завершают формирование цитозольной тирозинкиназы. В данном гене уже зафиксировано 341 уникальное молекулярное событие. В дополнение к мутациям, было обнаружено большое число вариантов или полиморфизмов.
Агаммаглобулинемия Брутона. Причины
Мутации в гене, лежащие в основе болезни Брутона, мешают развитию и функционированию В-лимфоцитов и их потомству. Основная идея заключается в том, что у здорового человека пре-В клетки созревают в лимфоциты. А у лиц страдающих этой болезнью, пре-В-клетки находятся или в малом количестве, или у них могут быть проблемы в функциональности.
Агаммаглобулинемия Брутона. Патофизиология
При отсутствии нормального белка, В-лимфоциты не дифференцируются или не созревают полностью. Без зрелых В-лимфоцитов, продуцирующие антитела плазмоциты также будут отсутствовать. Как следствие, ретикулоэндотелиальные и лимфоидные органы, в которых эти клетки пролиферируются, дифференцируются и хранятся, развиты плохо. Селезенка, миндалины, аденоиды, кишечник и периферические лимфоузлы, все они могут быть уменьшены в размере или вообще могут отсутствовать у лиц с Х-хромосомной агаммаглобулинемией.
Мутации в каждой из области гена могут привести к этой болезни. Самым распространенным генетическим событием является миссенс мутация. Большинство мутаций приводит к усечению белка. Эти мутации затрагивают критические остатки в цитоплазматическом белке и они весьма разнообразны и равномерно распределяются по всей молекуле. Тем не менее, тяжесть заболевания не может быть предсказана с помощью конкретных мутаций. Примерно одна треть точечных мутаций влияет на CGG сайты, которые, как правило содержат код для остатков аргинина.
Данный важный белок является необходимым для пролиферации и дифференцировки В-лимфоцитов. Мужчины с белковыми аномалиями имеют полное или почти полное отсутствие лимфоцитов в плазматических клетках.
Агаммаглобулинемия Брутона. Симптомы и проявления
Рецидивирующие инфекции начинают развиваться в раннем детстве и сохраняются на протяжении всей взрослой жизни.
Самым частым проявлением болезни Брутона или агаммаглобулинемии Брутона является увеличение восприимчивости к инкапсулированным гнойным бактериям, к таким как гемофильные инфекции, и некоторые виды Pseudomonas. Кожные инфекции у пациентов с болезнью в основном вызываются стрептококками группы А и стафилококками, они могут проявляться в виде импетиго, целлюлита, абсцессов, или фурункулов.
Форма экземы, которая напоминает атопический дерматит может быть очевидной, наряду с увеличением числа случаев гангренозной пиодермии, витилиго, алопеции и синдрома Стивенса-Джонсона (из-за более широкого использования препаратов). Другие инфекции, которые обычно присутствуют при этой болезни, включают энтеровирусные инфекции, сепсис, менингит и бактериальный понос. Пациенты также могут иметь аутоиммунные заболевания, тромбоцитопении, нейтропении, гемолитические анемии и ревматоидный артрит. Постоянные энтеровирусные инфекции очень редко приводят к смертельному энцефалиту или синдрому дерматомиозит-менингоэнцефалит. В дополнение к неврологическим изменениям, клинические проявления этого синдрома включают отеки и эритематозную сыпь на коже над разгибательными суставами.
Мужчины, могут развивать необычно тяжелый и / или периодический средний отит и пневмонии. Наиболее распространенным патогеном является S пневмония, а затем вирус гриппа B, стафилококки, менингококки и моракселла катаралис.
У детей в возрасте до 12 лет, типичные инфекции вызываются инкапсулированными бактериями. Общие инфекции в этой возрастной группе включают рецидивирующие пневмонии, синусит, и средний отит, которые вызываются S пневмония и вирусом гриппа В, которые трудно поддаются лечению в таком возрасте.
В зрелом возрасте, кожные проявления становятся более распространенными, как правило, из-за стафилококка и стрептококка группы А. Средний отит заменяется хроническим синуситом, и болезнь легких становится постоянной проблемой, как в ограничительной форме так и в обструктивной форме.
Как младенцы так и взрослые могут иметь аутоиммунные заболевания. Как правило, эти расстройства включают артрит, аутоиммунные гемолитические анемии, аутоиммунную тромбоцитопению, аутоиммунные нейтропении и воспалительные заболевания кишечника. Воспалительные заболевания кишечника могут быть очень трудно контролируемыми и они часто способствуют развитию хронической потери веса и недоеданию. Диарея является общей и вызывается Giardia или Campylobacter видами. Пациенты склонны к энтеровирусным инфекциям, в том числе к полиовирусу.
Физический осмотр
Младенцы мужского пола с агаммаглобулинемией Брутона, могут быть физически меньше, чем младенцы мужского пола без болезни из-за замедленного роста и развития от рецидивирующих инфекций.
При осмотре лимфатические узлы, миндалины, а также другие лимфоидные ткани могут быть очень маленькими или они вообще могут отсутствовать.
Заболевание диагностируется тогда, когда ребенку неоднократно становится плохо в присутствии различных инфекций, отита или стафилококковой инфекции кожи и конъюнктивита, которые не реагируют на терапию антибиотиками. Эти тяжелые инфекции могут быть связаны с нейтропенией.
Гангренозная пиодермия, например в виде язв и целлюлита нижних конечностей может также рассматриваться у некоторых пациентов.
Агаммаглобулинемия Брутона. Диагностика
Раннее выявление и диагностика имеет важное значение для предотвращения ранней заболеваемости и смерти от системных и легочных инфекций. Диагноз подтверждается аномально низкими уровнями или вообще отсутствующими зрелыми В-лимфоцитами, а также низкой или отсутствующей экспрессией тяжелой цепи μ на поверхности лимфоцитов. С другой стороны, уровень Т-лимфоцитов будет повышенным. Окончательный определитель болезни – молекулярный анализ. Молекулярный анализ также используется для пренатальной диагностики, которая может быть выполнена с помощью отбора проб ворсинок хориона или амниоцентеза, когда мать, как известно, является носителем дефектного гена. Уровни IgG менее 100 мг/дл подтверждают диагноз.
Редко, но диагноз может быть поставлен у взрослых в их втором десятилетии жизни. Это, как полагают, происходит из-за мутации в белке, а не из-за его полного отсутствия.
Лабораторные анализы
На первом этапе необходимо провести количественное измерение IgG, IgM, иммуноглобулина Е (IgE) и иммуноглобулин А (IgA). Уровни IgG следует измерять во-первых, желательно после возраста 6 месяцев, когда уровни материнских IgG начнут снижаться. Во-вторых, уровни IgG ниже 100 мг/дл, как правило, свидетельствует о болезни Брутона. Как правило, IgM и IgA не обнаруживаются.
После того, как уровень антител будет определен как аномально низкий, подтверждение диагноза будет достигнуто с помощью анализа B-лимфоцитных и Т-лимфоцитных маркеров. Уровни CD19 + В-клеток ниже 100 мг/дл. Значения анализа Т-клеток (CD4 + и CD8 +), как правило, увеличиваются.
Дальнейший анализ может быть проведен путем обнаружения ответов IgG к Т-зависимым и Т-независимым антигенам, путем проведения иммунизации, например после введения неконъюгированной 23-валентной пневмококковой или вакцин от дифтерии, столбняка и H гриппа типа B.
Молекулярно-генетическим исследованием можно установить раннее подтверждение диагноза врожденной агаммаглобулинемии.
Другие тесты
Исследования функций легких занимают центральное место в мониторинге заболеваний легких. Они должны проводиться ежегодно у детей, которые могут выполнять тест (обычно от 5 лет).
Процедуры
Эндоскопия и колоноскопия могут быть использованы для оценки масштабов и прогрессирования воспалительного заболевания кишечника. Бронхоскопия может быть полезной в диагностике и отслеживании хронического заболевания легких и инфекций.
Агаммаглобулинемия Брутона. Лечение
Лечебной терапии этой болезни не существует. Введение иммуноглобулина является основным методом контроля болезни. Типичные дозы – 400-600 мг/кг/мес должны назначаться каждые 3-4 недели. Дозы и интервалы могут быть скорректированы на основе отдельных клинических реакций. Терапия должна начинаться в возрасте 10-12 недель. Терапия с применением IgG должна начинаться с минимального уровня 500-800 мг/дл. Терапия должна начинаться в возрасте 10-12 недель.
Цефтриаксон может быть использован для лечения хронических инфекций, пневмонии, или сепсиса. Если это возможно, врачи должны получить культуры для выяснения чувствительности антибиотиков, так многие организмы уже сейчас проявляют устойчивость к многим антибиотикам. Инфекции стрептококка, в частности, могут потребовать цефтриаксона, цефотаксима или ванкомицина.
Бронхорасширители, стероидные ингаляторы, а также регулярные исследования функций легких (по крайней мере 3-4 раза в год) могут быть необходимой частью терапии в дополнение к антибиотикам.
Хронические дерматологические проявления атопического дерматита и экземы контролируются ежедневным увлажнением кожи специальными лосьонами и стероидами.
Хирургия
Хирургическое вмешательство может быть ограничено тяжелыми острыми инфекциями. Наиболее распространенные процедуры включают те, которые применяются для лечения пациентов с рецидивирующим отитом и с хроническим синуситом.
Агаммаглобулинемия Брутона. Осложнения
Осложнения включают хронические инфекции, энтеровирусные инфекции центральной нервной системы, увеличение частоты развития аутоиммунных заболеваний, а также инфекции кожи. Пациенты имеют повышенный риск развития лимфомы.
Агаммаглобулинемия Брутона. Прогноз
Большинство пациентов могут доживать до конца четвертого десятилетия своей жизни. Прогноз хороший, пока пациенты диагностируются и начинают лечиться рано регулярной внутривенной гамма-глобулин терапией.
Серьезные энтеровирусные инфекции и хронические легочные заболевания часто заканчиваются смертельным исходом в зрелом возрасте.
Синдром Ду́нкана (Да́нкана) - иммунодефицит, характеризующийся повышенной чувствительностью к вирусу Эпштейна-Барр. Ген повышенной чувствительности к вирусу локализован в Х-хромосоме, тип наследования заболевания рецессивный, поэтому болеют мальчики. У больных, перенёсших инфекционный мононуклеоз, развиваются длительное лихорадочное состояние, лимфаденопатия (увеличение лимфатических узлов), лимфоцитоз периферической крови, гепато- и спленомегалия. Позднее формируется В-клеточная лимфома, чаще в терминальных отделах тонкой кишки, от которой больные и погибают. Летальные исходы обусловлены также деструктивным гепатитом, вызываемым вирусом Эпштейна-Барр.
Недостаточность пурин-нуклеозид-фосфорилазы.
Недостаточность пурин-нуклеозид-фосфорилазы (ПНФ) наследуется по аутосомно-рецессивному типу. Дети страдают гипопластической анемией и крайне сниженной функцией Т-клеток.
Оротацидурия.
Оротацидури́я - наследственное заболевание синтеза пиримидинов, которое проявляется повышенной экскрецией оротовой кислоты (оротата) с мочой, недостаточностью Т-лимфоцитов, мегалобластной анемией и задержкой умственного и физического развития. При этом заболевании снижена активность ферментов оротидил-пирофосфорилазы и оротидил-декарбоксилазы, которые преобразуют оротовую кислоту в нуклеотид-оротидин-монофосфат, необходимый для синтеза нуклеиновых кислот.
Биотин-зависимые ферментопатии.
Биотин-зависимые ферментопатии также сопровождаются развитием клеточного иммунодефицита (наследственные дефекты биотинидазы и биотин-зависимых энзимов пируват-карбоксилазы и пропионат-карбоксилазы, участвующих в метаболизме аминокислот с разветвлённой цепью - валина, лейцина, изолейцина). Заболевание проявляется уже в периоде новорождённости эпизодами кетоацидоза, неврологической симптоматикой, алопецией, кожными сыпями и непереносимостью белка (рвота, мальдигестия, дегидратация). В моче содержится большое количество органических кислот. Дети отстают в физическом развитии. Из инфекционных процессов наиболее часто развиваются кандидоз и кератоконъюнктивиты. Биотин даёт хороший терапевтический эффект.
Первичные дефициты гуморального иммунитета.
К первичным дефицитам гуморального иммунитета относятся следующие основные синдромы:
Синдром Брутона (агаммаглобулинемия брутоновского типа)
Синдром Веста (недостаточность IgА)
Недостаточность IgG
Недостаточность транскобаламина II
Гипер-IgM-синдром
Гипер-IgЕ-синдром (синдром Джоба, синдром золотистого стафилококка с гипер-IgЕ)
Гипер-IgD-синдром (синдром ван дер Меера)
Поздний иммунный старт.
Наследственные дефициты гуморального иммунитета, за исключением синдрома Брутона и позднего иммунного старта, объединяют общим термином дисгаммаглобулинемии . При некоторых формах дисгаммаглобулинемий определяется нормальный или даже повышенный уровень иммуноглобулинов в крови и секретах слизистых оболочек.
Синдром Брутона.
Синдром Бру́тона (агаммаглобулинемия бру́тоновского типа) - недостаточность иммуноглобулинов всех классов. Заболевание было первым изученным иммунодефицитом наследственной природы (O. G. Bruton, 1952). Тип наследования - рецессивный, сцепленный с Х-хромосомой. В первые годы жизни развиваются инфекционные осложнения, преимущественно бактериальные. В то же время вирусные инфекции протекают у больных, как правило, легко. Первые признаки иммунодефицита становятся заметны на втором году жизни, хотя рецидивирующие инфекции могут появиться и у 8-месячного грудного младенца, и у 3-летнего ребёнка. Примерно у трети больных развивается вялотекущий артрит, похожий на ревматоидный, со стерильным выпотом в полость одного из крупных суставов.
В том случае, если заместительная терапия (введение препаратов иммуноглобулинов) начата прежде, чем повторные инфекции вызовут серьёзные морфологические изменения (например, бронхоэктазы, хроническую пневмонию и дыхательную недостаточность), ближайший прогноз очень хороший. Однако в подростковом и юношеском возрасте нередко развивается постепенно прогрессирующее неврологическое заболевание, напоминающее медленную вирусную инфекцию и проявляющееся дерматомиозитоподобным синдромом с выраженными отёками и периваскулярными лимфогистиоплазмоцитарными инфильтратами. Это тяжёлое системное заболевание приводит к летальному исходу. Считается, что оно вызвано энтеровирусной инфекцией (из крови и ликвора больных и умерших неоднократно выделяли энтеровирусы). В целом необходимо отметить высокую чувствительность этих больных к энтеровирусам. Так, дети с синдромом Брутона чаще болеют полиомиелитом, и он протекает у них тяжелее.
Болезнь Брутона – явление довольно редкое, но, тем не менее встречающееся. Это заболевание основано на генетической предрасположенности, то есть при недостатке выработки организмом антител, способных сопротивляться вирусам.
Немного о патологии
Эта патология – иммунодефицит, переданный по наследству и вызванный мутационными изменениями в генах для кодирования тирозинкиназы Брутона или обмена внутриклеточного сигнала. Эта болезнь в свое время правильно сформулирована ученым в 52 году прошедшего столетия, а сам ген и был назван в его честь.
Молекулы участвуют в созревании и обмене энергии на межклеточном уровне. Ген был найден на Х-хромосоме, кодирующей более 500 аминокислот, необходимых для окончательного формирования тирозинкиназы.
Мутационные изменения болезни не дают возможности развиваться и в дальнейшем функционировать В-лимфоцитам, назначение которых – это выработка антител и клеток памяти. Здорового человека отличает то, что эти клетки перерастают в B–лимфоциты, а у больных людей их количество небольшое и они менее активны.
Такие органы, как селезенка, аденоиды, кишечник, лимфоузлы и миндалины у больных этой патологией имеют маленькие параметры размеров либо отсутствовать могут полностью. Гипогаммаглобулинемия – эта патология вызвана недостатком клеток B-лимфоцитов в соотношении с уменьшением размера и количества антител.

Симптомы заболевания
Инфекции, провоцирующие это заболевание, могут начать свое развитие с раннего возраста и сохраняться на прежнем уровне всю жизнь. Агаммаглобулинемия Брутона проявляется в уязвимости организма к вирусным заболеваниям, в их числе гнойные воспалительные процессы, гемофилия и синегнойная палочка.
Поражения кожных покровов спровоцировано стрептококками группы A и стафилококками. Проявления на эпидермисе может быть в виде абсцесса, фурункул и целлюлита. Экзема напоминает аллергические высыпания на коже.
Иные инфекционные заболевания охватывают такие проявления, как бактериальная диарея, менингит и сепсис. Больные могут быть поражены аутоиммунными наследственными патологиями, артритами и тромбоцитопенией.
Регулярная подверженность пациента инфицированию может привести к менингоэнцефалиту или энцефалиту, который впоследствии приводят к летальному исходу. А также на теле появляется отечность и кожные высыпания в местах разгибания суставов.

Симптоматика по возрасту
У представителей сильного пола могут развиваться такие заболевания:
- последняя стадия отита;
- воспаление легких;
- вирус гриппа B;
- менингококки и стафилококки.
У детей в возрастной категории до 12 лет по причине этой патологии развивается бактериоз, заключенный в отдельные капсулы. Инфицирование, полученное от внешних негативных факторов, развивает средний отит, пневмонию, синусит и вирус гриппа В. Все эти полученные заболевания плохо поддаются лечению.
Во взрослом возрасте проблемы, связанные с высыпаниями на коже остаются надолго из-за постоянного питания стафилококковой или стрептококковой инфекциями, а средний отит постепенно перерастает в хронический синусит.
И маленькие дети и люди в любом возрасте могут быть поражены аутоиммунными заболеваниями.
Данные на основании осмотра специалистами показывают, что малыши мужского пола имеют небольшие параметры веса и роста, из-за того, что они не могут развиваться по причине болезни Брутона. Лимфоузлы или миндалины при осмотре могут не определяться вообще, или быть крайне маленькими.
Сама патология может быть выявлена только тогда, когда наступит ухудшение самочувствия ребенка, то есть он заболеет вирусным заболеванием и никакие медицинские препараты, в их числе антибиотики, не смогут помочь. Но также не исключено и развитие гангрены в виде язв на коже и наличии целлюлита на нижних конечностях.
Клиническая картина заболевания
После рождения у ребенка патология ни в чем не проявляется, так как содержание иммуноглобулина находится на нормальном уровне. Но на 3–5 месяце жизни, возможно, возникновение сепсиса или пиодермии, которые не поддаются лечению антибиотиками. Далее, болезнь поражает легкие, среднее ухо и желудочно-кишечный тракт. Отмечается наличие таких патологий, как менингит, остеомиелит и пансинусит.
Диагностика патологии
Ранее выявление болезни Брутона поможет избежать ее дальнейшего развития и летальных исходов от инфекций и легочных заболеваний. Сам факт патологии подтверждается отсутствием или очень низким уровнем лимфоцитов B, то при этом высоким уровнем лимфоцитов T.
Все это определяется на основании молекулярного анализа, который можно сделать на стадии беременности у матери – носителя такого гена. Анализ на иммуноглобулины, показывающий менее 100 единиц указывает на подтверждение этого заболевания. Иногда болезнь Брутона обнаруживается после 20 лет, потому что в белке произошла мутация.

Лабораторные анализы включают в себя:
- Проведение измерений количественных показателей иммуноглобулина Е и А, тестирование на антитела, причем последние лучше всего измерять по достижению 6 месяцев в период снижения материнских антител. В случае обнаружения менее 100 единиц этих показателей, это означает, что болезнь Брутона присутствует.
- После определения запредельно низкого уровня антител необходимо выполнить подтверждение такого выявления значения. В случае если белок, расположенный на поверхности В-лимфоцитов тоже ниже 100 единиц, но значение по анализу лимфоцитов Т-клеток увеличиваются.
- Далее, следует анализ, необходимый для определения чувствительности к вакцинам, например, пневмококковой.
Такими способами можно удостовериться о наличии заболевания Брутона.
Наряду с основными проводимыми исследованиями, следует постоянно держать на контроле состояние легких, как правило, это выполняется для детей в возрасте от 5 лет.

Лечение болезни
Для поддержания жизненных функций организма необходима терапия на протяжении всей жизни. Как правило, применяется внутривенная вакцинация иммуноглобулином или нативной плазмой, взятой от здоровых доноров.
При признании патологии в первый раз, проводиться заместительное лечение для насыщения до нормального уровня иммуноглобулина свыше 400 единиц. Если в это время у больного не наблюдается воспалительных и гнойных процессов, то можно далее продолжать ставить эту вакцину в качестве профилактики.
Если есть такое заболевание, как гнойнй абсцесс, независимо от места его расположения, требуется проведение лечения с помощью антибиотиков.
При лечении симптоматики заболевания выполняется промывание носовых пазух обеззараживающими средствами, вибрационный массаж грудной клетки и дренаж постуральный легких.

Прогнозы патологии
Если болезнь Брутона обнаружена у человека в раннем возрасте, до наступления более тяжелого ее проявления, то правильно назначенная и своевременно выполняемая терапия поможет сохранить нормальную жизнедеятельность.
Но, тем не менее статистикой подтверждается, что множество случаев заболевания обнаруживаются поздно в период воспалительных процессов, то такое обстоятельство грозит неблагоприятному дальнейшему развитию патологии.
Профилактические мероприятия
Это заболевание имеет генетическое происхождение, поэтому любые профилактические меры воздействия здесь бессильны. Чтобы не допустить проявление патологии, семейным парам пред рождением ребенка следует провериться и получить консультацию специалиста. Если же у новорожденного имеются признаки этого заболевания, то следует выполнять следующее:
- проведение терапевтических мероприятий;
- грамотно назначенная терапия;
- проведение вакцинации инактивированными препаратами.
ГЛАВА 3. ИММУНОДЕФИЦИТНЫЕ СОСТОЯНИЯ
Определение, клинические проявления и классификация иммунодефицитных состояний. Классификации первичных ИДС. Первичные ИДС адаптивного иммунитета с преимущественным поражением В-клеточного звена. Первичные ИДС адаптивного иммунитета с преимущественным поражением Т-клеточного звена. Комбинированные Т- и В-иммунодефициты. Первичные дефициты фагоцитарного звена врожденного иммунитета. Первичные дефициты системы комплемента. Предварительная диагностика первичных ИДС. Определение, основные причины формирования вторичных иммунодефицитов. Физиологические (возрастные) иммунодефициты. Роль предшествующих заболеваний, ятрогенных факторов, неблагоприятных экологических факторов и факторов риска образа жизни в возникновении вторичных ИДС.
В функционировании иммунной системы, как и любой другой системы организма, могут возникать нарушения, которые ведут к развитию заболеваний, характерных прежде всего для этой системы. Одним из самых распространенных вариантов таких нарушений являются иммунодефицитные состояния (ИДС), характеризующиеся неспособностью иммунной системы развивать нормальный иммунный ответ.
Иммунодефицитные состояния (ИДС) – это снижение функциональной активности основных компонентов иммунной системы, ведущее к нарушению иммунологического реагирования, к снижению или к формированию неадекватного врожденного или адаптивного иммунного ответа на самые различные патогены и антигены, в первую очередь – на инфекционные.
Необходимо отличать понятие иммунодефицит от понятия иммунологической толерантности, при которой также снижен или отсутствует иммунный ответ. В отличие от иммунодефицита, иммунологическая толерантность всегда специфична, то есть она формируется только против одного конкретного антигена или группы антигенов. При иммунодефицитных состояниях, как правило, снижается иммунологическая реактивность в целом.
Наличие ИДС в первую очередь приводит к снижению противоинфекционной защиты организма, что клинически проявляется в повышении инфекционной заболеваемости.
Для ИДС с преимущественным поражением В-клеточного звена адаптивного иммунитета характерно возникновение рецидивирующих инфекций, в том числе вызванных стафилококками, стрептококками, пневмококками, возбудителями кишечных инфекций и др.
При ИДС с преимущественным поражением Т-клеточного звена адаптивного иммунитета в дополнение ко многим бактериальным инфекциям возникают инфекционные заболевания, вызванные такой условно-патогенной флорой, как грибы Candida, вирусы (герпес, цитомегаловирус, аденовирусы), пневмоцисты, внутриклеточные бактерии (микобактерии, уреаплазмы и др.). Характерна также склонность к генерализации туберкулезной инфекции, повышена частота гельминтозов.
Первичные ИДС могут быть также обусловлены дефектами компонентов врожденного иммунитета. Эти дефекты могут затрагивать самые разные звенья системы врожденного иммунитета: компоненты и ингибиторы системы комплемента, дефекты фагоцитарных клеток, NK-клеток, рецепторов и транспортных молекул (ТАР), участвующих в процессинге антигенов. В связи с интенсивными исследованиями в этой области, которые проводятся с применением современных генетических и молекулярно-биологических методов, выявляются все новые варианты первичных ИДС врожденного иммунитета, а также сочетанных ИДС врожденного и адаптивного иммунитета. Как правило, первичные ИДС врожденного иммунитета проявляются в виде самых различных инфекционных заболеваний, например:
· Дефицит компонентов комплемента (С1, С4, С2; С3, С5-9 и др.) приводит к рецидивирующим генерализованным инфекциям, вызванным инкапсулированными микроорганизмами (Neisseria meningitidis, пневмококк, гемофильная палочка). Часто встречается системная красная волчанка.
· Дефекты фагоцитарной системы клинически чаще проявляются в виде инфекционных поражений кожи и паренхиматозных органов, например, вызываемых стафилококками и клебсиеллами при хронической гранулематозной болезни.
К клиническим проявлениям ИДС относятся также гематологические нарушения (лимфоаденопатии, лейкозы), характерно возникновение поражений желудочно-кишечного тракта, обусловленных в значительной мере нарушением местного иммунитета ЖКТ. Часто ИДС сопровождаются возникновением аутоиммунных и аллергических заболеваний. Особое место принадлежит онкологическим заболеваниям. Чаще всего у больных с ИДС встречаются гемобластозы, лимфопролиферативные заболевания, саркома Капоши.
Все иммунодефициты принято разделять на первичные, или врожденные, и вторичные, или приобретенные. Вторичные ИДС не являются генетически детерминированными, такие дефекты иммунной системы приобретаются в течение жизни и являются следствием каких-либо повреждающих воздействий на иммунную систему или наличия других заболеваний.
Первичные иммунодефицитные состояния
Первичные ИДС – это наследственные нарушения врожденной и/или адаптивной иммунной системы, связанные с генетическими дефектами одного или нескольких компонентов. Первичные ИДС адаптивной иммунной системы обусловлены преимущественными дефектами ее В-клеточного и Т-клеточного звена, во многих случаях имеют место сочетанные поражения обоих звеньев. Наиболее изученными первичными ИДС врожденного иммунитета являются ИДС, обусловленные генетическими дефектами системы комплемента и фагоцитарной системы.
При нормальном течении беременности во внутриутробном периоде развития ребенок находится в стерильных условиях. Немедленно после рождения его организм начинает заселяться микроорганизмами. Так как окружающая человека микрофлора не является патогенной, эта колонизация не вызывает болезни. В последующем встреча с патогенными микроорганизмами, с которыми ребенок еще не встречался, вызывает развитие соответствующей инфекционной болезни. Каждый контакт с патогеном приводит к расширению иммунологической памяти и формирует долговременный иммунитет.
В защите организма от постоянных атак патогенных вирусов, бактерий, грибов и простейших участвуют два основных компонента врожденной иммунной защиты: фагоцитоз и комплемент, а также два компонента адаптивной защиты: В-клеточный (антитела) и Т-клеточный. Каждый из этих компонентов может функционировать независимо, но чаще всего они работают в тесном комплексном взаимодействии. Врожденные дефекты одного из указанных звеньев приводят к нарушению защиты организма в целом и клинически проявляются в виде различных вариантов первичных иммунодефицитов.
В настоящее время, благодаря идентификации молекулярных нарушений, лежащих в основе патогенеза многих первичных иммунодфицитов, и в связи с большой вариабельностью клинической картины стало ясно, что этот вид патологии гораздо более распространен, чем считалось ранее.
Ранняя диагностика и адекватное лечение являются условием жизни пациентов с многими формами первичных иммунодефицитов. Без своевременной правильной диагностики и адекватного лечения ребенок погибает, как правило, в течение первого года жизни. Каждый вид иммунодефицита имеет свою, уже отработанную тактику лечения, и большинство первичных дефектов иммунной системы в настоящее время подлежит коррекции. Своевременная диагностика и лечение первичных иммунодефицитов являются одним из неиспользованных резервов для снижения заболеваемости, смертности и инвалидности населения в целом.
Первичные ИДС, как правило, имеют наследственную природу, поэтому их еще называют генетически обусловленными ИДС. Чаще всего первичные ИДС наследуются по рецессивному типу . В тех случаях, когда генетические дефекты затрагивают специфические факторы иммунитета (антителообразование, клеточные формы адаптивного иммунного ответа), их называют еще специфическими ИДС, в отличие от дефектов неспецифических (врожденных) компонентов защиты (фагоцитоза, системы комплемента и др.).
Иногда по отношению к первичным ИДС применяют термин «врожденные», что подразумевает возможность возникновения заболевания на основе наследственных дефектов, возникших в эмбриональном периоде, например, селективный дефицит IgA как следствие внутриутробно перенесенной коревой краснухи.
Первичные иммунодефициты разнообразны, и их описание и классификация в литературе постоянно меняются. Одной из первых классификаций иммунодефицитов адаптивной иммунной системы является предложенная в 1974 году Ю.М. Лопухиным и Р.В. Петровым классификация, в основу которой положены уровни генетических блоков на различных этапах дифференцировки Т- и В-лимфоцитов.
Согласно этой классификации, все многообразие форм ИДС адаптивной иммунной системы разделено на три группы:
1. Первичные ИДС с преимущественным поражением Т-системы (блок II, VI).
2. Первичные ИДС с преимущественным поражением B-системы (блок III, IV, V).
3. Комбинированные первичные ИДС с одновременным поражением Т-и В-систем (блок I, блоки V+VI и др.).
 I
I
Стволовая III
кроветворная
|
|
|
Рисунок 42. Патогенетическая схема классификации первичных ИДС Т- и В-систем иммунитета
(Р.В. Петров, Ю.М. Лопухин)
Блок I. Практически отсутствуют стволовые кроветворные клетки. Характерна генерализованная аплазия кроветворной и лимфоидной ткани. Не образуются ни Т-, ни В-лимфоциты.
К этому блоку относили тяжелую комбинированную иммунологическую недостаточность (ТКИН), которая встречается в различных формах.
Блок II. Блок на ранних этапах развития внутритимусных Т-лимфоцитов. Полное выключение клеточного адаптивного иммунитета. У таких больных характерно возникновение тяжелых рецидивирующих вирусных инфекций, приводящих к гибели в раннем (до 1 года) возрасте; высока частота врожденных аномалий развития; в 100-1000 раз выше риск возникновения злокачественных опухолей; организм пациента не отторгает чужеродный трансплантат.
Этот блок представлен, в частности, аплазией или гипоплазией тимуса (синдром Ди Джорджи).
Блок III. Для этого блока характерна агаммаглобулинемия, сцепленная с Х-хромосомой (болеют мальчики). У таких детей антитела практически не вырабатываются, что приводит в первую очередь к частым, тяжело протекающим бактериальным инфекциям. Этот вариант иммунодефицита называется болезнью Брутона, он диагносцируется, как правило, во втором полугодии жизни, так как в течение первых месяцев организм ребенка защищен материнскими антителами, прошедшими через плаценту и содержащимися в материнском молоке.
Блок IV. Несколько снижено число В-лимфоцитов. Гипогаммаглобулинемия с макроглобулинемией, так как синтез IgM сохранен или даже повышен, тогда как уровень IgG и IgA резко снижен.
Блок V. Селективный дефицит IgA. Из-за отсутствия основного секреторного иммуноглобулина у таких детей развиваются в первую очередь инфекционные заболевания, связанные со слизистыми оболочками носоглотки, дыхательных путей, ЖКТ, урогенитального тракта.
Блок VI. Затрагивает процессы созревания и выхода Т-лимфоцитов в периферические лимфоидные органы и кровяное русло. Характерна сочетанная патология с преимущественным поражением функций Т-клеточного иммунитета.
В настоящее время идентифицировано более 70 врожденных дефектов как врожденной, так и адаптивной иммунной системы. Вероятно, по мере совершенствования методов молекулярной иммунодиагностики, их число будет расти. Эндогенные, как правило, генетически обусловленные дефекты одного из компонентов иммунной системы приводят к нарушению всей системы защиты организма и клинически выявляются как одна из форм первичного иммунодефицитного состояния. Так как при нормальном функционировании иммунной системы в комплексном иммунном ответе участвуют многие типы клеток и сотни молекул, в основе патогенеза клинических форм первичных ИДС лежат многочисленные варианты дефектов.
В последние годы пришло осознание того факта, что клинические проявления первичных ИДС могут наблюдаться не только у новорожденных, но и в более позднем возрасте. Это связано с тем, что дефект одного из звеньев иммунитета может какое-то время не проявляться клинически в виде повышенной инфекционной заболеваемости, так как все другие компоненты иммунитета сохранены и компенсируют этот дефект пока не истощены их резервные возможности.
Первичные ИДС – это относительно редкие заболевания, их частота в среднем составляет 1/25000 - 1/100000, хотя селективный дефицит IgA встречается гораздо чаще: 1/500 - 1/700 человек. По данным Европейского регистра первичных ИДС за 1997 год частота зарегистрированных первичных ИДС в среднем по Европе составляла 1/96000 населения, тогда как в отдельных странах она была существенно выше: 1/38000 (Великобритания); 1/12500 (Швейцария); 1/10000 (Швеция). Скорее всего такие различия обусловлены уровнем развития медицины в разных странах, и реальная частота первичных ИДС существенно выше публикуемых средних цифр.
В настоящее время общепринятой является следующая упрощенная классификация первичных ИДС:
I. Первичные ИДС с преимущественным поражением гуморального адаптивного иммунитета
1. Сцепленная с Х-хромосомой агамма(гипогамма) глобулинемия – болезнь Брутона.
2. Общая вариабельная иммунологическая недостаточность (ОВИН) – общая вариабельная гипогаммаглобулинемия.
3. Транзиторная гипогаммаглобулинемия у детей (медленный иммунологический старт).
4. Избирательный дефицит иммуноглобулинов (селективный дефицит IgA).
II. Первичные ИДС с преимущественным поражением Т-клеточного адаптивного иммунитета
1. Синдром Ди Джорджи (гипо-, аплазия тимуса).
2. Хронический слизисто-кожный кандидоз.
III. Комбинированные Т- и В-иммунодефициты адаптивного иммунитета
1. Тяжелая комбинированная иммунологическая недостаточность (ТКИН):
а) Х-сцепленная;
б) аутосомно-рецессивные.
2. Атаксия – телеангиоэктазия (синдром Луи-Барр).
3. Синдром Вискотта-Олдрича.
4. Иммунодефицит с повышенным уровнем IgM (сцепленный с Х-хромосомой).
5. Иммунодефицит с карликовостью.
IV. Дефициты фагоцитарной системы
1. Хронический лимфогранулематоз.
2. Синдром Чедиака-Хигасси.
3. Синдром гипер-IgE (синдром Джоба).
V. Дефициты системы комплемента
1. Первичные ИДС, обусловленные дефицитом компонентов комплемента.
2. Первичные ИДС, обусловленные дефицитом инактиваторов системы комплемента.
Первичные ИДС с преимущественным поражением
гуморального адаптивного иммунитета
Болезнь Брутона
Впервые в 1952 году американский педиатр Брутон (Bruton) описал 8-летнего мальчика, страдавшего различными инфекционными заболеваниями, который к 4-летнему возрасту 14 раз болел пневмонией, неоднократно отитами, синуситами, перенес сепсис и менингит. При исследовании у него в сыворотке крови не обнаружили антител. Это было первое описание первичного иммунодефицита в научной медицинской литературе, выделенного как самостоятельная нозологическая форма.
Болезнь Брутона имеет рецессивный тип наследования, сцепленный с Х-хромосомой, болеют ей только мальчики. Заболевание также обозначается как Х-сцепленная агаммаглобулинемия. Встречается с частотой 1/1000000 населения. В Великобритании частота этого иммунодефицита составляет 1/100000.
В основе болезни Брутона лежит дефект фермента цитоплазматической тирозинкиназы, который осуществляет передачу сигнала в ядро В-лимфоцитов для его активации с последующим превращением в плазматическую клетку. Тем самым становится невозможным синтез иммуноглобулинов.
Первые признаки заболевания проявляются начиная с 7-8 месяцев до 2-3 лет. Трансплацентарный перенос иммуноглобулинов обычно обеспечивает больных этой болезнью детей количеством антител, достаточным для того, чтобы избегать инфекций в течение нескольких первых месяцев жизни. В дальнейшем отсутствие синтеза антител приводит, как правило, к возникновению рецидивирующих бактериальных инфекций, преимущественно дыхательных путей и кожи. Обычно возбудителями являются стрептококки, стафилококки и грамотрицательные бактерии. Инфекции имеют тенденцию распространяться, что приводит к септицемии и менингиту. Однако у таких детей сохраняется устойчивость к вирусным инфекциям, так как клеточный иммунитет не нарушен.
При объективном осмотре находят необычно маленькие, гладкие миндалины, лимфатические узлы небольшие, селезенка не увеличена. Обращает особое внимание тот факт, что лимфоузлы, печень и селезенка не реагируют увеличением на инфекционно-воспалительный процесс в организме, что является важным диагностическим признаком.
В слизистой оболочке кишечника полностью исчезают плазматические клетки, хотя в норме они там содержаться в достаточно большом количестве. В связи с этим наблюдаются нарушения всасывания, нередко развиваются хронические энтериты. В кишечнике часто выявляют абсцессы со скоплением распадающихся лейкоцитов в криптах кишки.
При лабораторном обследовании в периферической крови отмечается резкое снижение или отсутствие В-лимфоцитов, низкие уровни или отсутствие всех классов иммуноглобулинов.
Лечение: постоянная заместительная терапия иммуноглобулинами. Введение препаратов иммуноглобулинов производится внутривенно по 200-600 мг/кг в месяц. В случае возникновения инфекционного заболевания дополнительно вводят специфические иммуноглобулины (антистафилококковый, антистрептококковый, противокоревой и т.д.).
Прогноз: при правильно поставленном диагнозе и своевременно начатом лечении относительно благоприятный.