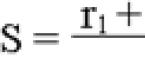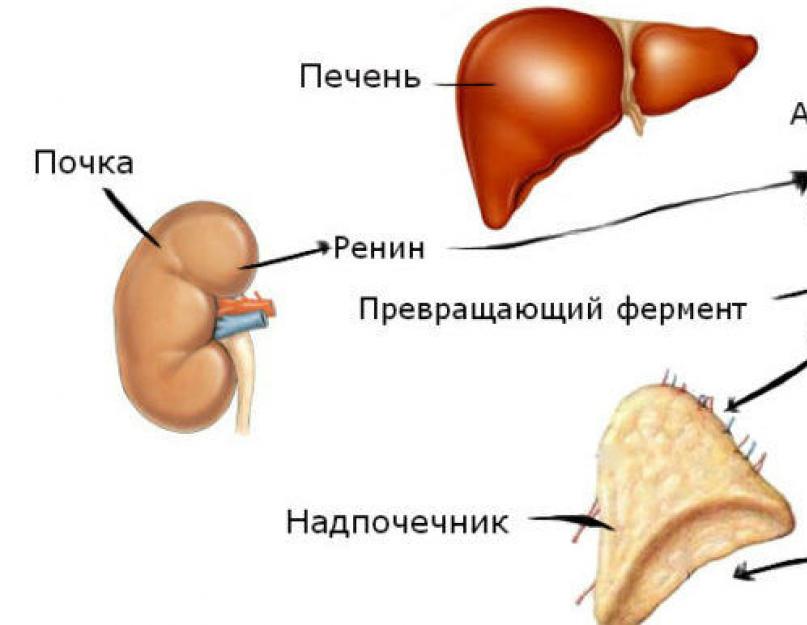
Ренин-ангиотензин-альдостероновая система (РААС) отвечает за норму объема экстрацеллюлярной жидкости, участвует в формировании стенок сосудов и обеспечивает уровень перфузии тканей. РААС непосредственно влияет на сердечно-сосудистую систему, нормализует артериальное давление и поддерживает содержание натрия и калия в норме.
В процессе участвует ренин (энзим), альдостерон (стероидный гормон) и ангиотензин II (пептидный гормон). Схема Ренин-ангиотензин-альдостероновой системы (РААС), представленная ниже, поможет понять принцип функционирования.
Основные цели РААС
Основной задачей для активации Ренин-ангиотензин-альдостероновой системы (РААС) является:
- Обеспечение достаточного кровотока в сосудах путем поддержания артериального давления, для функционирования печени, сердечно-сосудистой системы и сердца, почек, головного мозга.
- Выступает в роли «скорой помощи» при потере крови, при инфаркте и при резком снижении давления.
- Регулирует почечный и сосудистый гомеостаз, развивает процессы компенсаторного характера.
Длительная активация ренин-ангиотензин-альдостероновой системы может вызвать патологические явления в виде общего периферического сопротивления сосудов, недостаточный вывод жидкости из организма, избыток вырабатываемой крови, образованию периваскулярного и миокардиального фиброза.

Компонент системы ренин
Первым в звеньевой цепочке ренин-ангиотензин-альдостероновой системы, находится ренин, его производный элемент проренин, получается путем биосинтеза препрорениновой и рениновой РНК в юкстагломерулярных клетках. В дальнейшей подвергается глюкозелированию с последующим отщеплением аминокислот.
После деления, часть проренина выбрасывается в кровоток по принципу экзоцитоза, остаточная превращается в ренин, секретируясь юкстагломерулярными клетками аппарата почки, путем эндопептидаза. Образованный ренин в гранулах секреции юкстагломерулярной клетки в дальнейшем, также попадает в кровоток. Уровень производства ренина и дальнейшего поступления в кровь контролируется:
- артериальным давлением;
- химическими элементами NaClи Anq2;
- концентрацией внутри клетки ионов калия.

Ренин-ангиотензин-альдостероновая система призвана реагировать на сокращение объема воды и наличия натрия в организме при кровотечении. Потеря крови снижает давление в артериолах гломерулярных клубочков почек. Клетки стенок артериол улавливают спад натяжения, выделяют в капиллярную кровь ренин.
Большая часть регуляторов выработки ренина работает через почечные барорецепторы, под действием показателя состояния центральной нервной системы. На количество ренина влияет положение тела, переход из горизонтального положения в вертикальное или положение сидя, выработка энзима увеличивается. Это объясняется тем, что в симпатической части ЦНС повышается тонус и рефлекторно передается сигнал юкстагломерулярным клеткам.
В крови ренин воздействуя на ангиотензиноген, выделяет из него декапептид ангиотензин I, этот гормон не выполняет значимой функции в организме, но служит фундаментом для образования ангиотезина II. В процессе биохимической реакции, ангиотензин I, при помощи расщепления ангиотензинпревращающим ферментом (АПФ) переходит в ангиотезин II.
Ангиотезин II является центральным звеном ренин-ангиотензин-альдостероновой системы, основной задачей служит вазоконстрикторное воздействие на стенки артерий и ограниченное действие на центральную нервную систему. К рецепторам, участвующих в образование ангиотезина II, относятся следующие подтипы.
Агиотезин I-R (АТ 1-R) основа производного процесса, дает толчок основному количеству функций для реализации физиологически установленных норм ангиотезина II. Таким образом стимулируется выработка альдостерона надпочечниками, производится действие на симпатическую нервную систему. АТ 1-R мобилизирует ангиотезин II на рост клеток, и реагирование на воспалительный процесс. Влияние на сердечно-сосудистую систему, проявляется:
- повышением артериального давления;
- увеличением частоты сокращения сердечной мышцы;
- наличием сердечной и сосудистой гипертонии.

Следующий рецепторный тип АТ2-R к ангиотензину II, проявляет активность на первых стадиях развития эмбриона, при формировании мозга. На дальнейших этапах роста плода количество рецептора значительно сокращается.
Производный ангиотезина II – ангиотензиноген синтезируется печенью и под действием ренина, делится на ангиотезин I не активный декапептид, и на активный ангиотезин II, путем ферментативного воздействия АПФ. Функция активного октапептида ангиотезина II:
- путем сужения артериол повышает артериальное давление;
- контролирует выработку ренина юкстагломерулярными клетками;
- увеличивает сокращение миокарда;
- контролирует содержание натрия, ослаблением фильтрации в почках;
- поддержание водного баланса, путем формирования питьевого поведения.
Одной из важных задач ангиотезина II, воздействие на рецепторы центральной нервной системы, для активации биосинтеза в надпочечниках по выработке альдостерона. И путем обратной связи, всасывание ионов натрия почками.

Альдостерон
Синтез основного минералокортикоида происходит в гломерулярной зоне надпочечников, под воздействием калия и ангиотезина II и действует на мембранные рецепторы клеток ткани различных органов. Хотя основным производным альдостерона является ангиотезин II, сам гормон не участвует в производстве кортизола.
Функции альдостерона направлены на сдерживание натрия в почках и выведение лишнего количества натрия и калия из них. А также альдестерон играет немаловажную роль в ренин-ангиотензин-альдостероновой системе (РААС) отвечая:
- за защиту организма в неординарных ситуациях;
- стабилизирует уровень сахара в крови;
- сужение стенок сосуда, что делает невозможным понижение артериального давления, путем стабилизации кровяного потока.
Помимо регуляции артериального давления, альдостерон контролирует норму водно-солевого баланса. Но напрямую действуя на стенки сосудов, может вызвать нарушение функции эндотелия. Альдостерон способен спровоцировать воспаления стенки сосудов, активизировать моноциты крови и вызвать нарушение в почках и миокарде.
При повышенной выработке альдостерона или недостаточном количестве гормона необходимо медикаментозное лечение.
Catad_tema Артериальная гипертензия - статьи
Catad_tema Ожирение - статьи
Ожирение и артериальная гипертония
Опубликовано в журнале:ПРОБЛЕМЫ ЖЕНСКОГО ЗДОРОВЬЯ № 4, том 3, 2008
Е.И.Асташкин, М.Г.Глезер
Московская медицинская академия им. И.М.Сеченова
РЕЗЮМЕ
В обзоре анализируются роль ожирения в развитии артериальной гипертонии и сердечно-сосудистых заболеваний, патофизиологические механизмы этой связи, доминирующее значение ренин-ангиотензин-альдостероновой системы (РААС). Обсуждаются вопросы фармакологической коррекции высокого артериального давления у больных с ожирением с применением фиксированной комбинации препаратов, блокирующих РААС, и верапамила. Представлен анализ эффективности и безопасности применения сибутрамина для снижения веса у больных с высоким артериальным давлением.
Ключевые слова:
ожирение, артериальная гипертония, лечение.
ABSTRACT
Authors analyzed the role of obesity in development of arterial hypertension and cardiovascular diseases, pathophysiological mechanisms of this relationship, and dominating role of rennin-angiotensin-aldosterone system (RAAS). It was shown that pharmacological correction of high blood pressure in patients with obesity with fixed combination of RAAS blockers and verapamil is effective. The analysis of effectiveness and safety of sibutramine for weight loss in patients with high blood pressure is presented.
Key words:
obesity, arterial hypertension, treatment.
Актуальность рассматриваемой темы обусловлена тем, что во всем мире в последние годы наблюдается значительное увеличение числа людей, имеющих ожирение . Ожирение в настоящее время рассматривают как один из основных факторов, способствующих развитию заболеваний, которые являются главными причинами в структуре смертности среди взрослого населения. В первую очередь речь идет о развитии сахарного диабета 2 типа, а также сердечно-сосудистых и онкологических заболеваниях . Увеличение веса на 1 кг увеличивает риск сердечно-сосудистых заболеваний на 3,1% и диабета - на 4,5-9% .
Известно, что при ожирении риск развития артериальной гипертонии - фактора, также значительно влияющего на появление таких сердечно-сосудистых заболеваний, как инфаркты и инсульты, увеличен втрое по сравнению с людьми, имеющими нормальную массу тела. Как показано в исследовании INTERSALT, на каждые 4,5 кг прибавки веса систолическое артериальное давление (АД) увеличивается на 4,5 мм рт. ст. .
Ожирение, как фактор риска у женщин с артериальной гипертонией, особенно старшего возраста, встречается чаще, чем у мужчин. Одной из причин этого является гипоэстрогения, возникающая в период постменопаузы. Отмечают некоторые особенности распространенности ожирения при разных типах артериальной гипертонии. Так, среди пожилых женщин с изолированной систолической гипертонией ожирение встречается не столь часто, и нет данных о влиянии снижения веса на эту категорию пациентов . У женщин же с абдоминальным типом ожирения, имеющих систоло-диастолические формы артериальной гипертонии, снижение веса является важным моментом в контроле заболевания .
При ожирении возникает ряд гемодинамических изменений, в частности, увеличение объема циркулирующей крови, ударного объема и сердечного выброса при относительно нормальном сосудистом сопротивлении . Считается, что высокое АД у пациентов с ожирением обусловлено, главным образом, увеличенным сердечным выбросом при "неадекватно нормальном" периферическом сопротивлении .
Такое гемодинамическое состояние оказывает стимулирующее воздействие на две антагонистические регуляторные системы, контролирующие объем крови и периферическое сопротивление - ренин-ангиотензин-альдостероновую систему (РААС) и систему натриуретических пептидов сердца. Их нарушенная регуляция может в значительной степени объяснять высокий сердечный выброс у полных пациентов с артериальной гипертонией. Более того, эти сердечно-сосудистые регуляторные системы участвуют в метаболических изменениях, связанных с избыточной массой тела при сердечнососудистых заболеваниях .
Итак, при ожирении в патогенезе артериальной гипертонии играют существенную роль три основных механизма:
Патогенез развития артериальной гипертонии и сердечно-сосудистых заболеваний при ожирении схематично приведен на рисунке 1.
Рисунок 1. Схема патогенеза артериальной гипертонии и сердечно-сосудистых заболеваний при ожирении
Системная и тканевая ренин-ангиотензин-альдостероновая система и ее изменения при ожирении
В состав РААС входят ангиотензиноген, ренин, ангиотензин I, ангиотензин-превращающий фермент (АПФ) и ангиотензин II (АТ II). АТ II оказывает многообразное действие на разные клетки, имеющие специфические рецепторы.
Согласно классическим представлениям ангиотензиноген образуется в печени и под влиянием ренина, синтезируемого в околоклубочковых клетках почек (юкстагломерулярных клетках), ангиотензиноген превращается в крови в ангиотензин I. АПФ ответственен за расщепление АТ I, в результате которого образуется АТ II.
Важно отметить, что при ожирении происходит нарушение механизмов регуляции работы РААС . В физиологических условиях повышение активности РААС приводит к повышению сопротивления периферических сосудов и, соответственно, к повышению АД. По принципу обратной связи повышение АД должно вызывать снижение секреции ренина, падение уровня АТ II и уменьшать содержание альдостерона. Это, в свою очередь, снижает задержку жидкости и натрия и поддерживает АД на нормальном уровне.
Однако у пациентов с висцеральным ожирением нарушается регуляция уровня системных циркулирующих компонентов РААС . Несмотря на повышенное АД, задержку натрия и жидкости, а также увеличение объема циркулирующей крови, активность ренина плазмы и альдостерона остается нормальной или даже несколько повышенной . Подобное нарушение регуляции РААС при ожирении может быть следствием увеличения образования компонентов РААС и/или вторичного роста их концентрации, обусловленного дефектами в системе натрийуретических пептидов.
Было установлено, что помимо РААС крови существует тканевая, или так называемая локальная РААС, которая была выявлена в ряде тканей и органов, в том числе мозге, сердце, сосудах, почках, яичках, жировой ткани и др.
Как известно, ключевую роль в образовании АТ II играют два фактора: активность ренина и концентрация ангиотензиногена. Синтез и секреция ангиотензиногена в клетках разного типа не только определяет повышение локальной концентрации АТ II, но и увеличивает системную активность РААС. Хроническая инфузия АТ II мышам сопровождалась существенным увеличением содержания мРНК ангиотензиногена в адипоцитах . Эти результаты свидетельствуют о наличии положительной обратной связи между АТ II и ангиотензиногеном, когда увеличение уровня одного агента стимулирует образование второго. При ожирении, особенно висцерального типа, активность ренина в плазме сохраняется, как это уже указывалось, на нормальном или несколько повышенном уровне, а уровни ангиотензиногена и АТ II увеличены .
Структура и физиологические свойства жировой ткани
В состав жировой ткани входят клетки разного типа, в том числе адипоциты, макрофаги, фибробласты, эндотелиальные клетки сосудов и преадипоциты (адипобласты) . Последний вид клеток происходит из полипотентных стволовых клеток мезодермы. Из преадипоцитов во взрослом организме человека образуются новые дифференцированные ("малые") адипоциты . Эти адипоциты увеличиваются в размере ("большие" адипоциты) вследствие повышенного поступления с пищей жирных кислот. Длинноцепочечные жирные кислоты попадают в адипоциты из крови и депонируются в виде нейтральных триацилглицеринов. Жировая ткань ответственна за хранение и секрецию длинноцепочечных жирных кислот, выступающих в качестве одного из основных энергетических субстратов для многих органов и тканей, например, для сердечной и скелетной мускулатуры. "Большие" адипоциты секретируют существенно больше насыщенных жирных кислот. Гидролиз триглицеридов и высвобождение жирных кислот происходят под влиянием внутриклеточной гормон-чувствительной липазы, активность которой контролируется катехоламинами (положительная регуляция) и инсулином (отрицательная регуляция).
Эндокринная активность жировой ткани
В отличие от подкожного жира, который составляет обычно 75% от всей жировой ткани организма и является основным хранилищем липидов, висцеральный жир в настоящее время рассматривают как активную гормонпродуцирующую ткань.
Адипоциты продуцируют широкий спектр гормонов и цитокинов, участвующих в метаболизме глюкозы (адипонектин, резистин и др.), липидов (белок, переносящий эфиры холестерина), воспалении (ФНО-α, интерлейкин-6), коагуляции (ингибитор активатора плазминогена-1), регуляции давления крови (ангиотензиноген, АТ II), пищевом поведении (лептин), а также влияющих на метаболизм и функциональную активность различных органов и тканей, в том числе мышц, печени, мозга и сосудов (см. таблицу) .
Таблица. Эндокринная функция адипоцитов: адипоцитокины
| Адипоцитокины | Эффекты адипоцитокинов |
| Лептин | Поглощение пищи, масса жира |
| Адипонектин | |
| Резистин | Резистентность к инсулину, воспаление |
| Висфатин | Резистентность к инсулину |
| Оментин | Резистентность к инсулину |
| Серпин, высвобождающийся из жировой висцеральной ткани (Vaspin) | Резистентность к инсулину |
| Апелин | Вазодилатация |
| Белок, переносящий эфиры холестерина (CETP) | Метаболизм липидов |
| Липопротеиновая липаза (LPL) | Метаболизм липидов |
| Гормончувствительная липаза (HSL) | Метаболизм липидов |
| Белоксвязывающий жирные кислоты в адипоцитах-4 (A-FABP-4 (aP2)) | Метаболизм липидов |
| Перлипин | Метаболизм липидов |
| Ренитол связывающий белок (RBP) | Метаболизм липидов |
| Белокстимулирующий ацилирование (ASP) | Метаболизм липидов |
| Ангиотензин II (AT II) | Артериальное давление |
| Ангиотензин-превращающий фермент (ACE) | Артериальное давление |
| Ангиотензиноген (AGT) | Артериальное давление |
| Фактор некроза опухоли альфа (ФНО-а) | Воспаление |
| Интерлейкин, 6 (ИЛ-6) | Воспаление |
| С-реактивный белок (CRP) | Воспаление |
| Адипоцит-трипсин/комплемент фактор D (Адипсин) | Воспаление |
| Хемоаттрактантный белок макрофагов-1 (МСР-1) | Аттрактант для макрофагов |
| Межклеточная адгезионная молекула-1 (ICAM-1) | Активация макрофагов |
| Ингибитор активатора плазминогена-1 (PAI-1) | Фибринолиз |
Важно подчеркнуть, что даже небольшое увеличение объема висцерального жира играет значительную роль в нарушениях метаболизма, регуляции водноэлектролитного баланса и сердечно-сосудистых заболеваниях.
При увеличении массы жировой ткани содержание практически всех адипокинов в крови возрастает. Исключение составляет адипонектин, уровень которого в этих условиях падает . Лептин и адипонектин являются наиболее изученными адипокинами в настоящее время.
Лептин. Продукция лептина происходит, главным образом, в "больших" адипоцитах . Лептин часто рассматривается в качестве сигнальной молекулы, осуществляющей взаимосвязь между содержанием питательных веществ, поступающих в организм, состоянием жировой ткани и центральной нервной системой (гипоталамусом) . Лептин увеличивает окисление липидов в печени, а также липолиз в адипоцитах и скелетных мышцах . Инсулин стимулирует образование лептина. На уровень лептина также влияют свободные жирные кислоты, ФНО-α, эстрогены и гормон роста .
Адипонектин. Образование адипонектина происходит исключительно в адипоцитах . Адипонектин оказывает разнообразные биологические эффекты - оказывает антиатерогенное действие, увеличивает чувствительность клеток к инсулину, подавляет синтез глюкозы в печени, усиливает ее транспорт в мышцы, увеличивает окисление жирных кислот. Уровень адипонектина снижается при ожирении, резистентности к инсулину и сахарном диабете второго типа .
Жировая ткань и активность РААС
Оказалось, что жировая ткань занимает второе место после печени по образованию ангиотензиногена. Например, количество мРНК ангиотензиногена в адипоцитах составляет около 70% от уровня этого показателя в печени . Наличие взаимосвязи между уровнем ангиотензиногена, ожирением и артериальной гипертонией четко продемонстрировано в экспериментах на модели трансгенных мышей, экспрессирующих избыточные количества ангиотензиногена в жировой ткани. Эти мыши имеют висцеральное ожирение и гипертонию . Преадипоциты и дифференцированные жировые клетки имеют полный набор компонентов, необходимый для локального синтеза АТ II, а также АТ 1 рецептор для АТ II, что обеспечивает внутриклеточную передачу сигналов активации, запускаемых АТ II . При ожирении объем висцеральных дифференцированных адипоцитов увеличивается в 20-30 раз. Ожирение характеризуется дисфункцией адипоцитов, под которой понимают усиление образования и секреции различных адипокинов, цитокинов, а также увеличение содержания компонентов РААС, прежде всего, в висцеральном жире.
Суммируя данные различных исследований, можно констатировать, что при ожирении происходит повышение активности РААС, что находит свое отражение в следующих фактах:
Высокая активность РААС, в свою очередь, приводит к увеличению массы жировой ткани. В частности, трансгенные мыши, чрезмерно экспрессирующие ангиотензиноген только в жировых клетках, демонстрировали повышение уровня ангиотензиногена в крови, развитие гипертонии и увеличение массы жировой ткани . Тканевой АТ II по сути выполняет функцию фактора роста для адипоцитов . АТ II в результате воздействия на АТ 1 рецепторы вызывает увеличение белка циклина D 1 , который участвует в регуляции роста и деления жировых клеток . Показано, что АТ II индуцирует прохождение G 1 фазы клеточного цикла в преадипоцитах человека . Этот эффект был связан с влиянием на АТ 1 рецепторы и последующей активацией циклин D 1 -зависимой киназы .
Установлено, что АТ II вызывает дифференцировку преадипоцитов , активирует ключевые ферменты образования липидов (липогенеза) и увеличивает накопление триглицеридов в адипоцитах .
Висцеральное ожирение сопровождается увеличением активности 11-бета-гидроксистероид дегидрогеназы типа 1, что приводит к образованию кортизола, ключевого гормона дифференцировки преадипоцитов в адипоциты .
Активность тканевой РАС тесно связана с продукцией адипокинов жировой тканью. Показано, например, что АТ II вызывает экспрессию лептина в адипоцитах . Было высказано предположение, что такая активность свойственна только локально синтезируемому АТ II в отличие от системного АТ II .
Ожирение и активность симпатической нервной системы
При ожирении, особенно при абдоминальном его варианте, очень часто наблюдается активация симпатической нервной системы . В исследовании NAS (Normotesive Aging Study) было обнаружено увеличение норадреналина в моче, пропорциональное индексу массы тела . При снижении веса активность симпатической нервной системы уменьшается .
Повышению активности симпатической нервной системы при ожирении способствует наличие гиперинсулинемии и инсулинорезистентности. Инсулин может повышать активность симпатоадреналовой системы сам по себе, но отчасти это может быть связано с действием лептина. Известно, что по мере увеличения степени ожирения тощаковый уровень лептина, который секретируется адипоцитами, растет. Лептин увеличивает активность симпатической нервной системы, особенно в почках. Это приводит, с одной стороны, к высокому выбросу и увеличению частоты сердечных сокращений, а с другой - к повышению реабсорбции натрия и увеличению внутрисосудистого объема крови.
Установлено наличие взаимосвязи между РААС и симпатической нервной системой. С активацией симпатической нервной системы связывают усиление секреции ренина в почках, и происходит это независимо от внутрипочечной сенсорной системы, регулирующей секрецию ренина почками. Более того, увеличение циклического аденозинмонофосфата под влиянием катехоламинов, стимулирует экспрессию ангиотензиногена в адипоцитах человека . Увеличение уровня АТ II усиливает у людей активность симпатической нервной системы. Установлено, что АТ II активирует локальную симпатическую нервную систему, участвующую в повышении температуры тела (термогенезе). Холодовая обработка приводит к увеличению содержания АТ II в адипоцитах без сопутствующего изменения уровня АТ II в плазме .
Таким образом, нарушение регуляции РААС при ожирении также способно стимулировать активность симпатической нервной системы.
Методы фармакологической коррекции повышенного АД при ожирении
Вклад разных патогенетических механизмов в поддержание высокого АД при ожирении может быть различным. Следовательно, в этой ситуации благоприятное действие могут оказывать антигипертензивные препараты с самыми разными механизмами действия.
В соответствии с современными рекомендациями по лечению артериальной гипертонии залогом успеха значимого снижения АД является использование комбинированной терапии. Для пациентов с ожирением в первую очередь основные компоненты такой терапии должны содержать комбинацию препаратов, снижающих активность РААС (ИАПФ и сартаны), с препаратами, снижающими активность симпатической нервной системы (β-адреноблокаторы и недигидропиридиновые антагонисты кальция), и диуретиками. Высокая эффективность использования препаратов, блокирующих РААС, при ожирении показана во многих исследованиях . По поводу использования β-адреноблокаторов данные весьма противоречивы, прежде всего, вообще в связи с сомнениями в их полезности для лечения пациентов с неосложненной артериальной гипертонией, во-вторых, в связи с тем, что β-адреноблокаторы, во всяком случае классические, могут увеличивать вес пациентов и усиливать инсулинорезистентность . Следовательно, если и выбирать β-адреноблокаторы для лечения пациентов с ожирением или метаболическим синдромом, то это должны быть препараты, обладающие особыми свойствами, в частности, карведилол и небиволол.
В то же время установлено, что недигидропиридиновый антагонист кальция верапамил может не только значимо снижать АД , но и уменьшать активность симпатической нервной системы .
Таким образом, при ожирении для лечения артериальной гипертонии можно воспользоваться комбинацией препаратов, блокирующих РААС, и верапамила.
Следует подчеркнуть, что такого типа сочетание лекарственных препаратов существует в виде готовой комбинированной лекарственной формы - препарата Тарка, содержащего в своем составе жирорастворимый ИАПФ - трандолаприл и верапамил медленного высвобождении (верапамил СР). Такой подход очень важен для проведения эффективной терапии, так как использование готовых лекарственных форм улучшает приверженность пациентов к лечению .
Имеются данные, свидетельствующие о том, что препарат Тарка в большей степени, чем каждый из входящих в него компонентов, снижает АД, обладает выраженной способностью снижать гипертрофию левого желудочка, способствует нормализации эндотелиальной функции, является метаболически нейтральным, даже у пациентов с сахарным диабетом .
Двойное действие - снижение активности РААС под влиянием трандолаприла и симпатической нервной системы за счет верапамила пролонгированного действия - обеспечивают важное влияние на патогенетические механизмы развития артериальной гипертонии при ожирении и механизмы, провоцирующие поражение органов-мишеней при данном виде АГ.
Особое внимание, обсуждая лечение АГ при ожирении, следует обратить на то, что терапия, основанная на комбинации трандалаприла с верапамилом длительного действия, позволяет уменьшить риск развития сахарного диабета по сравнению с использованием другой тактики лечения - комбинации сартана с малой дозой тиазидного диуретика. Результаты исследования STAR отчетливо свидетельствует о том, что при применении препарата Тарка в течение одного года у меньшего числа людей с метаболическим синдромом, при котором абдоминальному ожирению придают главенствующее значение, развивается сахарный диабет (рис. 2) .
Рисунок 2. Развитие новых случаев сахарного диабета (глюкоза натощак > 126 мг/дл или 2-часовой уровень при проведении глюкозотолерантного теста > 200 мг/дл) в зависимости от типа антигипертензивной терапии у лиц с метаболическим синдромом в исследовании STAR

Кроме того по данным исследования SТAR-LET, даже при возникновении сахарного диабета на фоне лекарственной терапии перевод этих пациентов на прием препарата Тарка позволил у половины пациентов нормализовать углеводный обмен .
Результаты этих исследований заставляют пересмотреть рекомендации по медикаментозной терапии артериальной гипертонии у лиц с метаболическим синдромом и начинать терапию с комбинации, содержащей ИАПФ (или сартан) и антагонист кальция, или перевести пациентов на подобную терапию.
Как уже неоднократно упоминалось, для лечения артериальной гипертонии уменьшение веса пациентов и степени абдоминального ожирения играет важную роль. Конечно, снижение тем или иным способом массы тела может оказывать значимое влияние на снижение частоты сердечно-сосудистых заболеваний. В настоящее время существуют разные подходы для медикаментозной терапии ожирения . Первый - это симптоматическое лечение, а именно уменьшение количества потребляемых калорий за счет снижения всасывания жира, поступающего с пищей. Подобный подход можно назвать компенсаторным. Действительно, при такой терапии заболевание не устраняется (так как пациент продолжает переедать), а лишь временно компенсируется препаратом. Другой подход к лечению излишней массы тела и ожирения - это устранение сути проблемы, а именно хронического переедания. Так действует сибутрамин (препарат Меридиа). Он приводит к наступлению быстрого насыщения, снижает количество потребляемой пищи за счет подавления обратного захвата норадреналина и серотонина в синапсах нейрональных цепей . Сегодня Меридиа - это единственный оригинальный препарат, устраняющий причину ожирения.
Принципиальным отличием сибутрамина является то, что, не вызывая снижения аппетита, он способствует более раннему наступлению чувства сытости. Человек избавляется от патологической привычки переедать, результатом чего является постепенное и устойчивое снижение массы тела. Под влиянием сибутрамина потребление пищи снижается примерно на 20%. Наряду с этим сибутрамин опосредованно влияет на уровень биогенных аминов в крови, которые активируют адренорецепторы жировой ткани и инициируют липолиз в адипоцитах, что сопровождается изменением содержания энергетических субстратов в крови. Сибутрамин за счет активации β 2 - и β 3 -адренорецепторов усиливает процессы термогенеза и увеличивает расход энергии в организме.
Клиническая эффективность и безопасность сибутрамина (Меридиа) были продемонстрированы в большом количестве многоцентровых исследований. В частности, в исследовании STORM (Sibutramine Trial on Obesity Reduction and Maintenance), в которое включили 605 пациентов с ожирением, было показано, что двухлетний прием сибутрамина снижал вес пациентов в 3 раза, а окружность талии - в 2 раза более выраженно, чем плацебо . Важно, что в течение двух лет достигнутое снижение веса поддерживали 80% пациентов по сравнению с 16% пациентов, получавшими плацебо (p < 0,001). Показательно, что при этом улучшался липидный спектр: уровень липопротеидов высокой плотности повысился на 21% при снижении уровней липопротеидов низкой плотности и триглицеридов.
Положительное действие уменьшения веса в лечении пациентов с артериальной гипертонией и другими сердечно-сосудистыми заболеваниями может заключаться также в том, что уменьшение внутрибрюшного жира может снизить механическое сдавление почек, что может привести к улучшению их кровоснабжения и снижению активности РААС. Уменьшение жировой ткани внутри и вокруг почек может привести к снижению интерстициального давления, компрессии тонкой части петли Генли, увеличению кровотока в vasa recta, снижению канальцевой реабсорбции Na + и воды . Тем самым снижение веса, обусловленное методами немедикаментозной или медикаментозной коррекции, может уменьшать высоту артериального давления.
Однако до последнего времени в реальной клинической практике сибутрамин применяли с осторжностью, опасаясь его возможного негативного влияния на показатели АД и ЧСС, что, в свою очередь, могло приводить, хоть и у небольшого числа пациентов, но к неприятным субъективным ощущениям. Для изучения влияния сибутрамина на сердечно-сосудистую систему и доказательства безопасности препарата у группы пациентов с повышенным риском сердечно-сосудистых заболеваний было инициировано крупномасштабное многоцентровое двойное слепое плацебо контролируемое международное исследование SCOUT (Sibutramine Cardiovascular OUTcomes), где наблюдались 10 742 пациентов, из которых 97% имели заболевания сердечно-сосудистой системы, 88 - артериальную гипертонию и 84% - сахарный диабет 2 типа. По результатам первого завершившегося этапа исследования было установлено, что назначение сибутрамина привело к достоверному (р < 0,001) уменьшению веса (медиана изменения составила 2,2 кг), окружности талии (на 2 см в равной степени выраженному у мужчин и женщин) и снижению АД систолического на 3,0 мм рт. ст. и диастолического - на 1,0 мм рт. ст. Частота сердечных сокращений увеличивалась в среднем на 1,5 удара в минуту. Увеличение АД и увеличение частоты пульса наблюдалось соответственно у 4,7 и 3,5% пациентов. Таким образом, в данном исследовании было показано, что даже у пациентов, относящихся к группам высокого риска, применение сибутрамина (препарата Меридиа) было высокоэффективным и безопасным . Дальнейший анализ данных исследования SCOUT позволил установить, что у пациентов с артериальной гипертонией снижение АД при приеме сибутрамина было более выраженным и составило в среднем для систолического АД -6,5 (-27,0; 8,0) мм рт. ст., а для диастолического -2,0 (-15,0; 8,0) мм рт. ст. (p < 0,001). Среди пациентов, у которых снижение веса не было выраженным, снижение АД было достоверным, но менее выраженным, чем у лиц с успешным снижением веса, и составило в среднем для систолического -3,5 (-26,0; 10,0) мм рт. ст. и -1,5 (-16,0; 9,0) мм рт. ст. для диастолического АД (p < 0,001). У лиц с нормальным АД было достоверное, но не выраженное увеличение АД - 1,5 (-15,0; 19,5) мм рт. ст. систолического и на 1,0 (-10,5; 13,0) мм рт. ст. диастолического АД (p < 0,001). Степень повышения АД при приеме сибутрамина уменьшалась в соответствии со степенью потери веса .
Возникает закономерный вопрос, как будет соотноситься тот или иной вид антигипертензивной терапии с лечением сибутрамином. Несколько исследований было проведено для того, чтобы получить ответ на этот вопрос. Так, например, было показано, что использование комбинированной лекарственной формы, содержащей верапамил 180 мг/трандолаприл 2 мг в сочетании с сибутрамином 10 мг, приводило в течение 6 месяцев к более выраженному снижению АД, чем проведение только антигипертензивной терапии - систолическое АД снизилось, соответственно, на 21,9 ± 8,1 против 15,9 ± 12,3 мм рт. ст. и диастолическое - на 15,7 ± 8,1 против 9,1 ± 9,9 мм рт. ст. (p = 0,03). Комбинированная терапия приводила также к более выраженному улучшению антропометрических показателей; достоверное (p <5) по сравнению с исходным уровнем снижение малых липопротеидов низкой плотности, С-реактивного белка и висфатина наблюдалось только в группе пациентов, получавших комбинированную терапию сибутрамином с антигипертензиным препаратом Тарка .
В проспективном, многоцентровом, плацебо-контролируемом, двойном слепом исследовании HOS (Hypertension-Obesity-Sibutramine) в течение 16 недель было проведено сопоставление проведения различных режимов антигипертензивной терапии (фелодипин 5 мг/рамиприл 5 мг (n = 57), верапамил 180 мг/трандолаприл 2 мг (n = 55), метопролол сукцинат 95 мг/гидрохлортиазид 12,5 мг n = 59) при назначении сибутрамина и плацебо . В этом исследовании было подтверждено, что сибутрамин может повышать АД. Поэтому, конечно, необходимо адекватное проведение антигипертензивной терапии в период применения сибутрамина у пациентов с артериальной гипертонией. Показано также, что при лечении комбинацией β-адреноблокатора и гидрохлортиазида положительные эффекты сибутрамина по снижению веса, окружности талии и влиянию на метаболический профиль были выражены существенно меньше, чем при сочетании комбинированной терапии ИАПФ и антагонистами кальция с сибутрамином. Это еще раз подтверждает необходимость тщательного выбора антигипертензивной терапии у пациентов с ожирением, особенно при проведении программ, направленных на снижение веса. И в заключение следует отметить, что с нашей точки зрения одной из значимых проблем, снижающих эффективность борьбы с ожирением, является то, что ни врачи, ни население не рассматривают ожирение как значимый фактор риска. Более того, пациенты часто не оценивают себя как людей, имеющих ожирение. Например, в исследовании ПОЛОНЕЗ по оценке врачей, основанной на вычислении ИМТ, ожирение и у мужчин, и у женщин регистрировалось в три раза чаще, чем по самооценке пациентов . Таким образом, должна быть усилена и проводиться разъяснительная работа среди населения о необходимости предупреждения увеличения массы тела, коррекция имеющегося ожирения и важности постоянной терапии артериальной гипертонии. ЛИТЕРАТУРА
1. Flegal K.M., Carroll M.D., Ogden C.K., Johnson C.L. Prevalence and trends in obesity among US adults, 1999-2000. JAMA 2002; 288: 1723-7.
2. Pi-Sunyer F.X. The epidemiology of central fat distribution in relation to disease. Nutr Rev 2004; 62(7): 120-6.
3. Arbeeny C.M. Addressing the unmet medical need for safe and effective weight loss therapies. Obes Res 2004; 12(8): 1191-6.
4. Stamler J., Rose G., Stamler R., et al. INTERSALT study findings. Public health and medical care implications. Hypertension 1989; 14(5): 570-7.
5. Jedrychowski W., Mroz E., Bojanczyk M., Jedrychowska I. Excessive weight and hypertension in the elderly - the results of the community study. Arch Gerontol Geriatr 1991; 13(1): 61-9.
6. Kanai H., Tokunaga K., Fujioka S., et al. Decrease in intra-abdominal visceral fat may reduce blood pressure in obese hypertensive women. Hypertension 1996; 27(1): 125-9.
7. Poirier P., Giles T.D., Bray G.A., et al. American Heart Association; Obesity Committee of the Council on Nutrition, Physical Activity, and Metabolism. Obesity and cardiovascular disease: pathophysiology, evaluation, and effect of weight loss: an update of the 1997 American Heart Association Scientific Statement on Obesity and Heart Disease from the Obesity Committee of the Council on Nutrition, Physical Activity, and Metabolism. Circulation 2006; 113: 898-918.
8. Alpert M.A. Obesity cardiomyopathy; pathophysiology and evolution of the clinical syndrome. Am J Med Sci 2001; 321: 225-36.
9. Aneja A., El-Atat F., McFarlane S.I., Sowers J.R. Hypertension and obesity. Recent Progr Horm Res 2004; 59: 169-205.
10. Engeli S., Sharma A.M. The renin-angiotensin system and natriuretic peptides in obesity-associated hypertension. J Mol Med 2001; 79: 21-9.
11. Lafontan M., Moro C., Sengenes C., et al. An unsuspected metabolic role for atrial natriuretic peptides: the control of lipolysis, lipid mobilization, and systemic nonesterified fatty acids levels in humans. Arterioscler Thromb Vasc Biol 2005; 25: 2032-42.
12. Hall J.E. The kidney, hypertension and obesity. Hypertension 2003; 41(3): 625-33.
13. Cooper R., McFarlane Anderson N., Bennet F.I., et al. ACE, angiotensinogen and obesity: a potential pathway leading to hypertension. J Hum Hypertens 1997; 11: 107-11.
14. Lu Н., Boustany-Kari C.M., Daugherty A., Cassis L.A. Angiotensin II increases adipose angiotensinogen expression. Am J Physiol Endocrinol Metab 2007; 292: 1280-7.
15. Engeli S., Negrel R., Sharma A.M. Physiology and pathophysiology of the adipose tissue renin-angiotensin system. Hypertension 2000; 35(6): 1270-7.
16. Otto T.C., Lane M.D. Adipose development: from stem cell to adipocyte. Crit Rev Biochem Mol Biol 2005; 40: 229-42.
17. Hajer G.R., van Haeften T.W., Visseren F.L.J. Adipose tissue dysfunction in obesity, diabetes, and vascular diseases. Eur Heart J 2008; 29: 2959-71.
18. Wannamethee S.G., Lowe G.D., Rumley A., et al. Adipokines and risk of type 2 diabetes in older men. Diabetes Core 2007; 30: 1200-5.
19. Chu N.F., Spiegelman D., Hotamisligil G.S., et al. Plasma insulin, leptin, and soluble TNF receptoirs levels in relation to obesity-related atherogenic and thrombogenic cardiovascular disease risk factors among men. Atheroslerosis 2001; 157: 495-503.
20. Skurk T., Alberti-Huber C., Herder C., Hauner H. Relationship between adipocyte size and adipokine expression and secretion. J Clin Endocrinol Metab 2007; 6(92): 1023-33.
21. Ran J., Hirano T, Fukui T., et al. Angiotensin II infusion decreases plasma adiponectin level via its type 1 receptor in rats: an implication for hypertension related insulin resistance. Metabolism 2006; 55: 478-88.
22. Considine R.V., Sinha M.K., Heiman M.L., et al. Serum immunoreactive-leptin concentration in normal-weight and obese human. N Engl J Med 1996; 334: 292-5.
23. Schwartz M.W., Woods S.C., Porte D. Jr., et al. Central nervous system control of food intake. Nature 2000; 404(6778): 661-71.
24. Cheung C.C., Clifton D.K., Steiner R.A. Proopiomelanocortin neurons are direct targets for leptrin in the hypothalamus. Endocrinology 1997; 138; 4489-92.
25. Long Y.C., Zierath J.R. AMP-activated protein kinase signaling in metabolic regulation. J Clin Invest 2006; 116: 1776-83.
26. Saladin R., De Vos P., Guerre-Millo M., et al. Transient increase in obese gene expression after food intake or insulin administration. Nature 1995; 377: 527-9.
27. Zhang H.H., Kumar S., Barnet A.H., Eggo M.C. Tumor necrosis factor-alpha exerts dual effects on huuman adipose leptin synthesis and release. Mol Cell Endocrinol 2000; 159: 79-88.
28. Lindsay R.S., Funahashi T., Hanson R.L., et al. Adiponectin and development of type 2 diabetes in the Pima Indian population. Lancet 2002; 360: 57-8.
29. Hajer G.R., van der Graaf Y., Olijhoek J.K., et al. Low plasma levels of adiponectin are associated with low risk for future cardiovascular events in patients with clinical evident vascular disease. Am Heart J 2007; 154(750): 1-7.
30. Massiera F., Bloch-Faure M., Ceiler D., et al. Adipose angiotensinogen is involved in adipose tissue growth and blood pressure regulation. FASEB J 2001; 15: 2727-9.
31. Paul M., Mehr A.P., Kreutz R. Physiology of local renin-angiotensin systems. Physiol Rev 2006; 86: 747-803.
32. Karlsson C., Lindell K., Ottosson M., et al. Human adipose tissue expresses angiotensinogen and enzymes required for its conversion to angiotensin II. J Clin Endocrinol Metab 1998; 83: 3925-9.
33. Gorzelniak K., Engeli S., Janke J., et al. Hormonal regulation of the human adipose-tissue renin-angiotensin system: relationship to obesity and hypertension. J Hypertens 2002; 20: 965- 73.
34. Engeli S., Gorzelniak K., Kreutz R., et al. Co-expression of renin-angiotensin system genes in human adipose tissue. J Hypertens 1999; 17: 555-60.
35. Achard V., Boullu-Ciocca S., Desbriere R., et al. Renin receptor expression in human adipose tissue. Am J Physiol Regul Integr Comp Physiol 2007; 292: 274-82.
36. Sarzani R., Savi F., Dessi-Fulgheri P., Rappelli A. Renin-angiotensin system, natrriuretic peptides, obesity, metabolic syndrome, and hypertension: an integrated view in human. J Hypertens 2008; 26: 831-43.
37. Crandall D.L., Armellino D.C., Busler D.E., et al. Angiotensin II receptors in human preadipocytes: role in cell cycle regulation. Endocrinology 1999; 140: 154-8.
38. Saint-Marc P., Kozak L.P., Ailhaud G., et al. Angiotensin II as a trophic factor of white adipose tissue: stimulation of adipose cell formation. Endocrinology 2001; 142: 487-92.
39. Watanabe G., Lee R.J., Albanese C., et al. Angiotensin II activation of cyclin D1-dependent kinase activity. J Biol Chem 1996; 271: 22570-7.
40. Darimont C., Vassaux G., Ailhaud G., Negrel R. Differentiation of preadipose cells: paracrine role of prostacyclin upon stimulation of adipose cells by angiotensin-II. Endocrinology 1994; 135: 2030-6.
41. Jones B.H., Standridge M.K., Moustaid N. Angiotensin II increases lipogenesis in 3T3-L1 and human adipose cells. Endocrinology 1997; 138: 1512-9.
42. Wake D.J., Walker B.R. Inhibition of 11beta-hydroxysteroid dehydrogenase type 1 in obesity. Endocrine 2006; 29: 101-8.
43. Engeli S., Bohnke J., Feldpausch M., et al. Regulation of 11beta-HSD genes in human adipose tissue: influence of central obesity and weight loss. Obes Res 2004; 12: 9-17.
44. Kim S., Whelan J., Claycombe K., et al. Angiotensin II increases leptin secretion by 3T3-L1 and human adipocytes via a prostaglandin-independent mechanism. J Nutr 2002; 132: 1135-40.
45. Cassis L.A., English V.L., Bharadwaj K., Boustany C.M. Differential effects of local versus systemic angiotensin II in the regulation of leptin release from adipocytes. Endocrinology 2004; 145: 169-74.
46. Mancia G., Bousquet P., Elghozi J.L., et al. The sympathetic nervous system and the metabolic syndrome. J Hypertens 2007; 25: 909-20.
47. Tentolouris N., Liatis S., Katsilambros N. Sympathetic system activity in obesity and metabolic syndrome. Ann N Y Acad Sci 2006; 1083: 129-52.
48. Landsberg L., Troisi R., Parker D., et al. Obesity, blood pressure, and the sympathetic nervous system. Ann Epidemiol 1991; 1: 295-303.
49. Serazin V., Dos Santos E., Morot M., Giudicelli Y. Human adipose angiotensinogen gene expression and secretion are stimulated by cyclic AMP via increased DNA cyclic AMP responsive element binding activity. Endocrine 2004; 25: 97-104.
50. Cassis L.A. Role of angiotensin II in brown adipose thermogenesis during cold acclimation. Am J Physiol Endocrinol Metab 1993; 265: 860-5.
51. Cassis L.A., Dwoskin L.P. Presynaptic modulation of neurotransmitter release by endogenous angiotensin II in brown adipose tissue. J Neural Transm 1991; 34: 129-37.
52. Reisin E., Weir M., Falkner B., et al. Lisinopril versus hydrochlorothiazide in obese hypertensive patients: a multi-center placebo-controlled trial. Treatment in Obese Patients With Hypertension (TROPHY) Study Group. Hypertension 1997; 30(1): 2140-5.
53. Neutel J.M., Saunders E., Bakris G.L., et al. The efficacy and safety of low- and high-dose fixed combinations of irbesartan/hydrochlorothiazide in patients with uncontrolled systolic blood pressure on monotherapy: the INCLUSIVE trial. J Clin Hypertens (Greenwich) 2005; 7(10): 578-86.
54. Беленков Ю.Н., Чазова И.Е., Мычка В.Б. от имени исследовательской группы "ЭКО". Многоцентровое рандомизированное открытое исследование по изучению эффективности изменения образа жизни и терапии ингибиторами АПФ (квинаприлом) у больных ожирением и артериальной гипертонией (ЭКО). Артериальная гипертензия 2003; 9(6): 3-6.
55. Jacob S., Rett K., Henriksen E.J. Antihypertensive therapy and insulin sensitivity: do we have to redefine the role of beta-blocking agents? Am J Hypertens 1998; 11(10): 1258-65.
56. Kaaja R., Kujala S., Manhem K., et al. Effects of sympatholytic therapy on insulin sensitivity indices in hypertensive postmenopausal women. Int J Clin Pharmacol Ther 2007; 45(7): 394-401.
57. Kuperstein R., Sasson Z. Effects of antihypertensive therapy on glucose and insulin metabolism and on left ventricular mass: a randomized, double-blind, controlled study of 21 obese hypertensives. Circulation 2000; 102(15): 1802-6.
58. Halperin A.K., Cubeddu L.X. The role of calcium channel blockers in the treatment of hypertension. Am Heart J 1986; 111(2): 363-82.
59. McAllister R.G. Jr. Clinical pharmacology of slow channel blocking agents. Prog Cardiovasc Dis 1982; 25(2): 83-102.
60. Binggeli C., Corti R., Sudano I., et al. Effects of chronic calcium channel blockade on sympathetic nerve activity in hypertension. Hypertension 2002; 39(4): 892-6.
61. Lefrandt J.D., Heitmann J., Sevre K., et al. The effects of dihydropyridine and phenylalkylamine calcium antagonist classes on autonomic function in hypertension: the VAMPHYRE study. Am J Hypertens 2001; 14(11): 1083-9.
62. Wanovich R., Kerrish P., Gerbino P.P., Shoheiber O. Compliance patterns of patients treated with 2 separate antihypertensive agents versus fixed-dose combination therapy Am J Hypertens 2004; 175: 223.
63. Dezii C.M. A retrospective study of persistence with single-pill combination therapy vs. concurrent two-pill therapy in patients with hypertension. Manag Care. 2000; 9(9): 2-6.
64. Gerbino P.P., Shoheiber O. Adherence patterns among patients treated with fixed-dose combination versus separate antihypertensive agents. Am J Health Syst Pharm 2007; 64(12): 1279-83.
65. Jackson K. Persistence of fixed versus free combination with valsartan and HCTZ for patients with hypertension. Value Health Suppl 2006; 9: 363.
66. Aepfelbacher F.C., Messerli F.H., Nunez E., Michalewicz L. Cardiovascular effects of a trandolapril/verapamil combination in patients with mild to moderate essential hypertension. Am J Cardiol 1997; 79(6): 826-8.
67. Reynolds N.A., Wagstaff A.J., Keam S.J. Trandolapril/verapamil sustained release: a review of its use in the treatment of essential hypertension. Drugs 2005; 65(13): 1893-914.
68. Sharma S.K., Ruggenenti P., Remuzzi G. Managing hypertension in diabetic patients - focus on trandolapril/verapamil combination. Vasc Health Risk Manag 2007; 3(4): 453-65.
69. Bakris G., Molitch M., Hewkin A., et al. Differences in glucose tolerance between fixed-dose antihypertensive drug combinations in people with metabolic syndrome. Diabetes Care 2006; 29(12): 2592-7.
70. Bakris G., Molitch M., Zhou Q., et al. Reversal of diuretic-associated impaired glucose tolerance and new-onset diabetes: results of the STAR-LET study. J Cardiometab Syndr 2008; 3(1): 18-25.
71. Bailey C.J., Day C. New pharmacological approaches to obesity. Obesity Practice 2005; 1: 2-5.
72. Yanovski S.Z., Yanovski J.A.Y. Obesity. N Engl J Med 2002; 346(8): 591-602.
73. Day C., Bailey C.J. Sibutramine update. Br J Diabet Vasc Dis 2002; 2: 392-7.
74. James W.P., Astrup A., Finer N., et al. Effect of sibutramine on weight maintenance after weight loss: a randomised trial. STORM Study Group. Sibutramine Trial of Obesity Reduction and Maintenance. Lancet 2000; 356: 2119-25.
75. Torp-Pedersen C., Caterson I., Coutinho W., et al. Cardiovascular responses to weight management and sibutramine in high-risk subjects: an analysis from the SCOUT trial. Eur Heart J 2007; 28(23): 2915-23.
76. Sharma A.M., Caterson I.D., Coutinho W., et al. Blood pressure changes associated with sibutramine and weight management - an analysis from the 6-week lead-in period of the sibutramine cardiovascular outcomes trial (SCOUT). Diabetes Obes Metab 2008. doi 10.1111/J.1463-1326.2008.00930.
77. Nakou E., Filippatos T.D., Liberopoulos E.N., et al. Effects of sibutramine plus verapamil sustained release/trandolapril combination on blood pressure and metabolic variables in obese hypertensive patients. Expert Opin Pharmacother 2008; 9(10): 1629-39.
78. Scholze J., Grimm E., Herrmann D., et al. Optimal treatment of obesity-related hypertension: the Hypertension-Obesity-Sibutramine (HOS) study. Circulation 2007; 115(15): 1991-8.
79. Глезер М.Г. Результаты российского исследования ПОЛОНЕЗ (Эффективность и безоПаснОсть энаренаЛа у пациентОв с артериальНой гипЕртенЗией). Терапевтический архив 2006; 4: 44-50.
Ренин
Ренин - протеолитический фермент, продуцируемый юкстагломерулярными клетками, расположенными вдоль афферентных (приносящих) артериол почечного тельца. Секрецию ренина стимулирует падение давления в приносящих артериолах клубочка, вызванное уменьшением АД и снижением концентрации Na + . Секрецию ренина также способствует снижение импульсации от барорецепторов предсердий и артерий в результате уменьшения АД. Секрецию ренина ингибирует Ангиотензин II, высокое АД.
В крови ренин действует на ангиотензиноген.
Ангиотензиноген - α 2 -глобулин, из 400 АК. Образование ангиотензиногена происходит в печени и стимулируется глюкокортикоидами и эстрогенами. Ренин гидролизует пептидную связь в молекуле ангиотензиногена, отщепляя от него N-концевой декапептид -ангиотензин I , не имеющий биологической активности.
Под действием антиотензин-превращающего фермента (АПФ) (карбоксидипептидилпептидазы) эдотелиальных клеток, лёгких и плазмы крови, с С-конца ангиотензина I удаляются 2 АК и образуется ангиотензин II (октапептид).
Ангиотензин II
Ангиотензин II функционирует через инозитолтрифосфатную систему клеток клубочковой зоны коры надпочечников и ГМК. Ангиотензин II стимулирует синтез и секрецию альдостерона клетками клубочковой зоны коры надпочечников. Высокие концентрации ангиотензина II вызывают сильное сужение сосудов периферических артерий и повышают АД. Кроме этого, ангиотензин II стимулирует центр жажды в гипоталамусе и ингибирует секрецию ренина в почках.
Ангиотензин II под действием аминопептидаз гидролизуется в ангиотензин III (гептапептид, с активностью ангиотензина II, но имеющий в 4 раза более низкую концентрацию), который затем гидролизуется ангиотензиназами (протеазы) до АК.
Альдостерон
Альдостерон - активный минералокортикостероид, синтезирующийся клетками клу-бочковой зоны коры надпочечников.
Синтез и секрецию альдостерона стимулируют ангиотензин II , низкая концентрация Na + и высокая концентрацией К + в плазме крови, АКТГ, простагландины. Секрецию альдостерона тормозит низкая концентрация К + .
Рецепторы альдостерона локализованы как в ядре, так и в цитозоле клетки. Альдостерон индуцирует синтез: а) белков-транспортёров Na + , переносящих Na + из просвета канальца в эпителиальную клетку почечного канальца; б) Na + ,К + -АТФ-азы в) белков-транспортёров К + , переносящих К + из клеток почечного канальца в первичную мочу; г) митохондриальных ферментов ЦТК, в частности цитратсинтазы, стимулирующих образование молекул АТФ, необходимых для активного транспорта ионов.
В результате альдостерон стимулирует реабсорбцию Na + в почках, что вызывает задержку NaCl в организме и повышает осмотическое давление.
Альдостерон стимулирует секрецию К + , NH 4 + в почках, потовых железах, слизистой оболочке кишечника и слюнных железах.
3. Схема регуляции водно-солевого обмена Роль системы раас в развитии гипертонической болезни
Гиперпродукция гормонов РААС вызывает повышение объема циркулирующей жидкости, осмотического и артериального давления, и ведет к развитию гипертонической болезни.
Повышение ренина возникает, например, при атеросклерозе почечных артерий, который возникает у пожилых.
Гиперсекреция альдостерона – гиперальдостеронизм , возникает в результате нескольких причин.
Причиной первичного гиперальдостеронизма (синдром Конна ) примерно у 80% больных является аденома надпочечников, в остальных случаях - диффузная гипертрофия клеток клубочковой зоны, вырабатывающих альдостерон.
При первичном гиперальдостеронизме избыток альдостерона усиливает реабсорбцию Na + в почечных канальцах, что служит стимулом к секреции АДГ и задержке воды почками. Кроме того, усиливается выведение ионов К + ,Mg 2+ и Н + .
В результате развиваются: 1). гипернатриемия, вызывающая гипертонию, гиперволемию и отёки; 2). гипокалиемия, ведущая к мышечной слабости; 3). дефицит магния и 4). лёгкий метаболический алкалоз.
Вторичный гиперальдостеронизм встречается гораздо чаще, чем первичный. Он может быть связан с сердечной недостаточностью, хроническими заболеваниями почек, а также с опухолями, секретирующие ренин. У больных наблюдают повышенный уровень ренина, ангиотензина II и альдостерона. Клинические симптомы менее выражены, чем при первичном альдостеронизе.
КАЛЬЦИЙ, МАГНИЙ, ФОСФОРНЫЙ ОБМЕН
Функции кальция в организме:
Внутриклеточный посредник ряда гормонов (инозитолтрифосфатная система);
Участвует в генерации потенциалов действия в нервах и мышцах;
Участвует в свертывании крови;
Запускает мышечное сокращение, фагоцитоз, секрецию гормонов, нейромедиаторов и т.д.;
Участвует в митозе, апоптозе и некробиозе;
Увеличивает проницаемость мембраны клеток для ионов калия, влияет на натриевую проводимость клеток, на работу ионных насосов;
Кофермент некоторых ферментов;
Функции магния в организме:
Является коферментом многих ферментов (транскетолаз (ПФШ), глюкозо-6ф дегидрогеназы, 6-фосфоглюконат дегидрогеназы, глюконолактон гидролазы, аденилатциклазы и т.д.);
Неорганический компонент костей и зубов.
Функции фосфата в организме:
Неорганический компонент костей и зубов (гидроксиаппатит);
Входит в состав липидов (фосфолипиды, сфинголипиды);
Входит в состав нуклеотидов (ДНК, РНК, АТФ, ГТФ, ФМН, НАД, НАДФ и т.д.);
Обеспечивает энергетический обмен т.к. образует макроэргические связи (АТФ, креатинфосфат);
Входит в состав белков (фосфопротеины);
Входит в состав углеводов (глюкозо-6ф, фруктозо-6ф и т.д.);
Регулирует активность ферментов (реакции фосфорилирования / дефосфорилирования ферментов, входит в состав инозитолтрифосфата – компонента инозитолтрифосфатной системы);
Участвует в катаболизме веществ (реакция фосфоролиза);
Регулирует КОС т.к. образует фосфатный буфер. Нейтрализует и выводит протоны с мочой.
Распределение кальция, магния и фосфатов в организме
У взрослого человека содержится в среднем 1000г кальция:
Кости и зубы содержат 99% кальция. В костях 99% кальция находится в виде малорастворимого гидроксиапатита [Са 10 (РО 4) 6 (ОН) 2 Н 2 О], а 1% - в виде растворимых фосфатов;
Внеклеточная жидкость 1%. Кальций плазмы крови представлен в виде: а). свободных ионов Са 2+ (около 50%); б). ионов Са 2+ соединённых с белками, главным образом, с альбумином (45%); в) недиссоциирующих комплексов кальция с цитратом, сульфатом, фосфатом и карбонатом (5%). В плазме крови концентрация общего кальция составляет 2, 2-2,75 ммоль/л, а ионизированного - 1,0-1,15 ммоль/л;
Внутриклеточная жидкость содержит кальция в 10000-100000 раз меньше чем внеклеточной жидкости.
Во взрослом организме содержится в около 1кг фосфора:
Кости и зубы содержат 85% фосфора;
Внеклеточная жидкость – 1% фосфора. В сыворотке крови концентрация неорганического фосфора – 0,81-1,55 ммоль/л, фосфора фосфолипидов 1,5-2г/л;
Внутриклеточная жидкость – 14% фосфора.
Концентрация магния в плазме крови 0,7-1,2 ммоль/л.
Обмен кальция, магния и фосфатов в организме
С пищей в сутки должно поступать кальция - 0,7-0,8г, магния - 0,22-0,26г, фосфора – 0,7-0,8г. Кальций всасывается плохо на 30-50%, фосфор хорошо – на 90%.
Помимо ЖКТ, кальций, магний и фосфор поступают в плазму крови из костной ткани, в процессе ее резорбции. Обмен между плазмой крови и костной тканью по кальцию составляет 0,25-0,5г/сут, по фосфору – 0,15-0,3г/сут.
Выводится кальций, магний и фосфор из организма через почки с мочой, через ЖКТ с калом и через кожу с потом.
Регуляция обмена
Основными регуляторами обмена кальция, магния и фосфора являются паратгормон, кальцитриол и кальцитонин.
Паратгормон
Паратгормон (ПТГ) - полипептид, из 84 АК (около 9,5 кД), синтезируется в паращитовидных железах.
Секрецию паратгормона стимулирует низкая концентрация Са 2+ ,Mg 2+ и высокая концентрация фосфатов, ингибирует витамин Д 3 .
Скорость распада гормона уменьшается при низкой концентрации Са 2+ и увеличивается, если концентрация Са 2+ высока.
Паратгормон действует на кости и почки . Он стимулирует секрецию остеобластамиинсулиноподобного фактора роста 1 и цитокинов , которые повышают метаболическую активностьостеокластов . В остеокластах ускоряется образованиещелочной фосфатазы и коллагеназы , которые вызывают распад костного матрикса, в результате чего происходит мобилизация Са 2+ и фосфатов из кости во внеклеточную жидкость.
В почках паратгормон стимулирует реабсорбцию Са 2+ ,Mg 2+ в дистальных извитых канальцах и уменьшает реабсорбцию фосфатов.
Паратгормон индуцирует синтез кальцитриола (1,25(OH) 2 D 3).
В результате паратгормон в плазме крови повышает концентрацию Са 2+ иMg 2+ , и снижает концентрацию фосфатов.
Гиперпаратиреоз
При первичном гиперпаратиреозе (1:1000) нарушается механизм подавления секреции паратгормона в ответ на гиперкальциемию. Причинами могут быть опухоль (80%), диффузная гиперплазия или рак (менее 2%) паращитовидной железы.
Гиперпаратиреоз вызывает:
разрушение костей , при мобилизации из них кальция и фосфатов. Увеличивается риск переломов позвоночника, бедренных костей и костей предплечья;
гиперкальциемию , при усилении реабсорбции кальция в почках. Гиперкальциемия приводить к снижению нервно-мышечной возбудимости и мышечной гипотонии. У больных появляются общая и мышечная слабость, быстрая утомляемость и боли в отдельных группах мышц;
образования в почках камней при увеличение концентрации фосфата и Са 2+ в почечных канальцах;
гиперфосфатурию и гипофосфатемию , при снижении реабсорбции фосфатов в почках;
Вторичный гиперпаратиреоз возникает при хронической почечной недостаточности и дефиците витамина D 3 .
При почечной недостаточности угнетается образование кальцитриола, что нарушает всасывание кальция в кишечнике и приводит к гипокальциемии . Гиперпаратиреоз возникает в ответ на гипокальциемию, но паратгормон не способен нормализовать уровень кальция в плазме крови. Иногда возникает гиперфостатемия. В следствие повышения мобилизации кальция из костной ткани развивается остеопороз.
Гипопаратиреоз
Гипопаратиреоз обусловлен недостаточностью паращитовидных желёз и сопровождается гипокальциемией. Гипокальциемия вызывает повышение нервно-мышечной проводимости, приступы тонических судорог, судороги дыхательных мышц и диафрагмы, ларингоспазм.
Кальцитриол
Кальцитриол синтезируется из холестерола.

В коже под влиянием УФ-излучения из 7-дегидрохолестерола образуется большая часть холекальциферола (витамина Д 3). Небольшое количество витамина Д 3 поступает с пищей. Холекальциферол связывается со специфическим витамин Д-связывающим белком (транскальциферином), поступает в кровь и переносится в печень.
В печени 25-гидроксилаза гидроксилирует холекальциферол в кальцидиол (25-гидроксихолекальциферол, 25(OH)Д 3). D-связывающий белок транспортирует кальцидиол в почки.
В почках митохондриальная 1α-гидроксилаза гидроксилирует кальцидиол в кальцитриол (1,25(OH) 2 Д 3), активную форму витамина Д 3 . Индуцирует 1α-гидроксилазу паратгормон.
Синтез кальцитриола стимулирует паратгормон, низкая концентрация фосфатов и Са 2+ (через паратгормон) в крови.
Синтез кальцитриола ингибирует гиперкальциемия, она активирует 24α-гидроксилазу , которая превращает кальцидиол в неактивный метаболит 24,25(OH) 2 Д 3 , при этом соответственно активный кальцитриол не образуется.
Кальцитриол воздействует на тонкий кишечник, почки и кости.
Кальцитриол:
в клетках кишечника индуцирует синтез Са 2+ -переносящих белков, которые обеспечивают всасывание Са 2+ , Mg 2+ и фосфатов;
в дистальных канальцах почек стимулирует реабсорбцию Са 2+ , Mg 2+ и фосфатов;
при низком уровне Са 2+ увеличивает количество и активность остеокластов, что стимулирует остеолиз;
при низком уровне паратгормона, стимулирует остеогенез.
В результате кальцитриол повышает в плазме крови концентрацию Са 2+ , Mg 2+ и фосфатов.
При дефиците кальцитриола нарушается образование аморфного фосфата кальция и кристаллов гидроксиапатитов в костной ткани, что приводит к развитию рахита и остеомаляции.
Рахит - заболевание детского возраста, связанное недостаточной минерализацией костной ткани.
Причины рахита : недостаток витамина Д 3 , кальция и фосфора в пищевом рационе, нарушение всасывания витамина Д 3 в тонком кишечнике, снижением синтеза холекальциферола из-за дефицита солнечного света, дефект 1а-гидроксилазы, дефект рецепторов кальцитриола в клетках-мишенях. Снижение концентрации в плазме крови Са 2+ стимулирует секрецию паратгормона, который через остеолиз вызывает разрушение костной ткани.
При рахите поражаются кости черепа; грудная клетка вместе с грудиной выступает вперёд; деформируются трубчатые кости и суставы рук и ног; увеличивается и выпячивается живот; задерживается моторное развитие. Основные способы предупреждения рахита - правильное питание и достаточная инсоляция.
Кальцитонин
Кальцитонин - полипептид, состоит из 32 АК с одной дисульфидной связью, секретируется парафолликулярными К-клетками щитовидной железы или С-клетками паращитовидных желёз.
Секрецию кальцитонина стимулирует высокая концентрация Са 2+ и глюкагона, подавляет низкая концентрация Са 2+ .
Кальцитонин:
подавляет остеолиз (снижая активность остеокластов) и ингибирует высвобождение Са 2+ из кости;
в канальцах почек тормозит реабсорбцию Са 2+ , Mg 2+ и фосфатов;
тормозит пищеварение в ЖКТ,
Изменения уровня кальция, магния и фосфатов при различных патологиях
Снижение концентрации Са 2+
беременности;
алиментарной дистрофии;
рахите у детей;
остром панкреатите;
закупорке желчевыводящих путей, стеаторее;
почечной недостаточности;
вливание цитратной крови;
Повышение концентрации Са 2+ в плазме крови наблюдается при:
переломы костей;
полиартриты;
множественные миеломы;
метастазы злокачественных опухолей в кости;
передозировка витамина Д и Са 2+ ;
механическая желтуха;
Снижение концентрации фосфатов в плазме крови наблюдается при:
гиперфункции паращитовидных желез;
остеомаляции;
почечный ацидоз
Повышение концентрации фосфатов в плазме крови наблюдается при:
гипофункции паращитовидных желез;
передозировка витамина Д;
почечной недостаточности;
диабетическом кетоацидозе;
миеломной болезни;
остеолизе.
Концентрация магния часто пропорциональна концентрации калия и зависит от общих причин.
Повышение концентрации Mg 2+ в плазме крови наблюдается при:
диабетическом ацидозе;
тиреотоксикозе;
хроническом алкоголизме.
распаде тканей;
инфекциях;
Роль микроэлементов: Mg 2+ , Mn 2+ , Co , Cu , Fe 2+ , Fe 3+ , Ni , Mo , Se , J . Значение церулоплазмина, болезнь Коновалова-Вильсона.
Марганец – кофактор аминоацил-тРНК синтетаз.
Биологическая роль Na + , Cl - , K + , HCO 3 - - основных электролитов, значение в регуляции КОС. Обмен и биологическая роль. Анионная разность и ее коррекция.
Тяжелые металлы (свинец, ртуть, медь, хром и др.), их токсическое действие.
Повышение содержание хлоридов в сыворотке крови : обезвоживание, острая почечная недостаточность, метаболический ацидоз после диареи и потери бикарбонатов, респираторный алкалоз, травма головы, гипофункция надпочечников, при длительном приеме кортикостероидов, тиазидный диуретиков, гиперальдостеронизм, болезнь Кушенга.
Снижение содержания хлоридов в сыворотке крови : алкалоз гипохлоремический (после рвоты), ацидоз респираторный, избыточное потоотделение, нефрит с потерей солей (нарушение реабсорбции), травма головы, состояние с увеличением объема внеклеточной жибкости, калит язвенный, болезнь Аддисона (гипоальдостеронизм).
Повышенное выделение хлоридов с мочой : гипоальдостеронизм (болезнь Аддисона), нефрит с потерей солей, повышенный прием соли, лечение диуретиками.
Снижение выведения хлоридов с мочой : Потеря хлоридов при рвоте, диареи, болезнь Кушинга, терминальная фаза почечной недостаточности, ретенция соли при образовании отеков.
Выделение кальция с мочой в норме 2,5-7,5 ммоль/сут.
Повышение содержание кальция в сыворотке крови : гиперпаратиреоз, метастазы опухолей в костную ткань, миеломная болезнь, сниженное выделение кальцитонина, передозировка витамина Д, тиреотоксикоз.
Снижение содержания кальция в сыворотке крови : гипопаратиреоз, увеличение выделения кальцитонина, гиповитаминоз Д, нарушение реабсорбции в почках, массивная гемотрансфузия, гипоальбунемия.
Повышенное выделение кальция с мочой : длительное воздействие солнечных лучей (гипервитаминоз Д), гиперпаратиреоз, метастазы опухолей в костную ткань, нарушение реабсорбции в почках, тиреотоксикоз, остеопороз, лечение глюкокортикоидами.
Снижение выведения кальция с мочой : гипопаратиреоз, рахит, острый нефрит (нарушение фильтрации в почках), гипотериоз.
Повышение содержание железа в сыворотке крови : апластическая и гемолитическая анемии, гемохроматоз, острый гепатит и стеатоз, цирроз печени, талассемия, повторные трансфузии.
Снижение содержания железа в сыворотке крови : железодефицитная анемия, острые и хронические инфекции, опухоли, заболевания почек, кровопотеря, беременность, нарушение всасывания железа в кишечнике.
Ренин
– фермент, синтезируемый юкстагломерулярными клетками почечных афферентных артериол, имеющий ММ около 40 кДа. Особенно интенсивно образование ренина происходит при ишемии почек. Локализация юкстагломерулярных клеток делает их особенно чувствительными к изменениям кровяного давления, а также концентрации ионов Na + и К + в жидкости, протекающей через почечные канальцы. Благодаря указанным свойствам любая комбинация факторов, вызывающая снижение объема жидкости (обезвоживание, падение кровяного давления, кровопотеря и др.) или снижение концентрации NaCl, стимулирует высвобождение ренина.
В то же время большинство регуляторов синтеза ренина действуют через почечные барорецепторы. На освобождение ренина оказывает влияние состояние ЦНС, а также изменение положения тела в пространстве. В частности, при переходе из положения лёжа в положение сидя или стоя (клиностатическая проба) секреция ренина увеличивается. Эта рефлекторная реакция обусловлена повышением тонуса симпатической части автономной нервной системы, передающей импульсы к b-адренорецепторам юкстагломерулярных клеток.
Основным субстратом, на который воздействует ренин, является ангиотензиноген – белок, входящий во фракцию a 2 -глобулинов и образуемый печенью. Под воздействием глюкокортикоидов и эстрогенов синтез ангиотензиногена значительно возрастает. В результете действия ренина ангиотензиноген превращается в декапептид ангиотензин I. Это соединение обладает чрезвычайно слабым действием и существенного влияния на уровень кровяного давления не оказывает.
Между тем ангиотензин I под воздействием так называемого ангиотензинпревращающего фермента (АПФ) переходит в мощный сосудосуживающий фактор – ангиотензин II. АПФ (дипептидкарбооксипептидаза) является интегральным белком, расположенным преимущественно на мембране эндотелиальных клеток, эпителии, мононуклеарах, нервных окончаниях, клетках репродуктивных органов и др. Растворимая форма АПФ присутствует практически во всех жидкостях организма.
Принято выделять две изоформы АПФ. Первая из них получила условное наименование «соматической». Эта изоформа имеет ММ 170 кДа и включает гомологичные С- и N-домены. Вторая форма АПФ («репродуктивная») найдена в семенной жидкости, имеет ММ около 100 кДа и соответствует С-домену первой изоформы АПФ. Каждый из 2 указанных доменов содержит аминокислотные остатки, которые могут принимать участие в образовании связи с атомом цинка. Такие Zn 2+ -структуры являются типичными для многих металлопротеиназ и оказываются основными участками взаимодействия фермента как с субстратом, так и с ингибиторами АПФ.
Следует заметить, что АПФ не только приводит к образованию ангиотензина II, но и разрушает брадикинин – соединение, расширяющее кровеносные сосуды. Следовательно, увеличение кровяного давления при воздействии АПФ связано как с образованием ангиотензина II, так и с распадом брадикинина (рис. 32).
Важную роль для действия АПФ играет ионный состав и, в частности, содержание ионов хлора. Так, при высокой концентрации Cl – С-домен АПФ гидролизует и брадикинин, и ангиотензин-I быстрее, чем N-домен. Во внеклеточных регионах, где концентрация анионов хлора высока, за превращение ангиотензина-I отвечает преимущественно N-домен. Однако внутриклеточно, где концентрация Cl – низкая, N-домен может участвовать в гидролизе других пептидных субстанций.
За последние годы установлено, что АПФ играет важную роль в гемопоэзе, ибо под его воздействием ингибируется образование гематопоэтического пептида , тормозящего образование гематопоэтических клеток костного мозга.
Роль АПФ в организме была выявлена на мышах, лишенных гена АПФ. У таких животных отмечалось низкое кровяное давление, различные сосудистые дисфункции, нарушение структуры и функции почек и бесплодие у самцов.
Ангиотензин II
увеличивает кровяное давление, вызывая сужение артериол, и является самым сильнодействующим из известных вазоактивных агентов. Кроме того, он по механизму обратной связи тормозит образование и высвобождение ренина юкстагломерулярными клетками почки, что в конечном итоге должно восстанавливать нормальный уровень кровяного давления. Под воздействием ангиотензина II резко возрастает продукция основного минералокортикоида – альдостерона. Несмотря на то, что это действие является прямым, ангиотензин II не влияет на продукцию кортизола. Основное назначение альдостерона сводится к задержке Na + (за счет усиления его реабсорбции в почечных канальцах) и выделению К + и Н + (главным образом через почки). Эти реакции осуществляются следующим образом.
Альдостерон
проникает из внеклеточной жидкости в цитоплазму клетки и там соединяется со специфическим рецептором, после чего образовавшийся комплекс (альдостерон+рецептор) проникает в ядро. Альдостерон также стимулирует открытие Na + каналов, благодаря чему ионы Na + входят в клетку через апикальную мембрану из просвета канальца.
Увеличение секреции К + под воздействием альдостерона обусловлено возрастанием проницаемости апикальной мембраны по отношению к этим ионам, благодаря чему К + поступает из клетки в просвет канальца.
Задержка Na + в организме, как и ангиотензин II, способствует повышению кровяного давления.
Ангиотензин II способен связываться со специфическими рецепторами клубочковых клеток надпочечника. Содержание этих рецепторов во многом зависит от концентрации ионов К + . Так, если уровень К + повышается, то возрастает число рецепторов к ангиотензину II в клубочковых клетках. При уменьшении концентрации ионов К + отмечается прямо противоположный эффект. Следовательно, ионы К + играют основную роль в действии ангиотензина II на надпочечники.
За последнее время установлено, что ангиотензин II способен активировать макрофаги, благодаря чему усиливается агрегация тромбоцитов и ускоряется свёртывание крови. Одновременно при этом высвобождается ингибитор активатора плазминогена- I (ИАП-1), что может сопровождаться депрессией фибринолиза. Ангиотензин II является одним из факторов, способствующих развитию атерогенеза, торможению апоптоза и усилению оксидативного стресса в тканях, тем самым провоцируя агрегацию тромбоцитов и тромбообразование.
Ангиотензин II способен усиливать функцию миокарда, участвует в биосинтезе норадреналина и других физиологически активных веществ. Одновременно он может действовать как ростовой фактор, приводя к сосудистой и сердечной гипертрофии.
У некоторых животных и у человека ангиотензин II под воздействием фермента аминопептидазы превращается в гептапептид ангиотензин III. У человека уровень ангиотензина II приблизительно в 4 раза выше, чем ангиотензина III. Оба эти соединения оказывают влияние на уровень кровяного давления и продукцию альдостерона и довольно быстро разрушаются под воздействием ферментов ангиотензиназ.
При тяжелых заболеваниях почек, сопровождающихся их ишемией, благодаря повышенному образованию и секреции ренина наблюдается стойкое повышение кровяного давления (почечная гипертензия). Применение ингибиторов АПФ в этих условиях приводит к быстрой нормализации кровяного давления.
В заключение следует еще раз подчеркнуть, что ангиотензин-ренино-альдостероновая система теснейшим образом связана с функцией калликреин-кининовой системы, ибо образование ангиотензина II и разрушение брадикинина осуществляется под воздействием одного и того же фермента – АПФ.
Ещё в конце ХІХ века стало известно, что почки принимают активное участие в регуляции артериального давления. Они вырабатывают фермент – ренин, который с ангиотензином и альдостероном составляет РААС (ренин-ангиотензин-альдостероновую систему). Они влияют на водно-солевой обмен, артериальное давление (именно поэтому различные патологии почек сопровождаются ) и выполняют другие функции.
Что такое ренин-ангиотензин-альдостероновая система
Принцип действия РААСКазалось бы, ренин – фермент, вырабатываемый почками, ангиотензиноген – гликопротеид, синтезируемый печенью, а альдостерон вообще гормон надпочечников – что между ними общего. Тем не менее, они составляют единую систему, которая запускается выработкой ренина в юкстагломерулярных клетках почек.
Существует несколько механизмов стимуляции синтеза фермента:
- Макулярный. Он срабатывает, если снижается поступление ионов натрия в дистальном извитом канальце.
- Внутрипочечный барорецепторный. Юкстагломерулярные клетки являются барорецепторами, они воспринимают растяжение стенок артериол, соответственно реагируют на снижение давления выработкой ренина.
- Симпатический. Юкстагломерулярные клетки иннервируются симпатической нервной системой, и как только к ним поступает сигнал, они тут же начинают синтезировать фермент, способствующий повышению давления. Именно поэтому при стрессах, психо-эмоциональных нагрузках возникает артериальная гипертензия.
Затем ренин поступает в кровь. Там он воздействует на гликопротеин ангиотензиноген, вырабатываемый печенью. Таким образом, ангиотензиноген превращается в ангиотензин І. Под влиянием ангиотензинпревращающего фермента (АПФ) отщепляется дипептид у ангиотензина І, и он становится самым мощным сосудосуживающим средством – ангиотензином ІІ. Кроме того, что он вызывает спазм гладкой мускулатуры, тормозит выработку брадикинина, он стимулирует синтез альдостерона. Этот гормон, вырабатываемый надпочечниками:
- удерживает ионы натрия и воду;
- выводит калий;
- усиливает синтез АТФ-азы воздействуя на ДНК.
Как только нормализуется концентрация натрия в крови, прекращается выработка ренина. Все продукты реакций распадаются, давление нормализуется, и начинают синтезироваться вазодилататоры:
- брадикинин;
- каллидин.
Стимулироваться работа ренин-ангиотензин-альдостероновой системы может из-за различных патологий. Например, при стенозе почечной артерии запускается РААС. Из-за того, что вырабатывается эффективнейший вазоконстриктор ангиотензин ІІ, возникает спазм сосудов. А это приводит к неоправданной гипертонии. Давление значительно повышается, соответственно нарушается микроциркуляция крови. К органам приносится меньшее количество питательных веществ, жизненно необходимых микроэлементов и кислорода (без него клетки мозга начинают отмирать через 5 минут).
Функции РААС
Как только в дистальных канальцах почек понижается концентрация ионов натрия, на юкстагломерулярные клетки подаётся сигнал от симпатической нервной системы, барорецепторы реагируют на расширение стенки артериол, тут же включается ренин-ангиотензин-альдостероновая система. Все реакции происходят практически мгновенно, но даже за столь незначительное время РААС справляется со своими функциями:
- поддерживает кислотно-щелочной баланс;
- регулирует водно-солевой обмен;
- влияет на восстановление объёма крови;
- усиливает скорость клубочковой фильтрации.
На протекание химических реакций влияет кислотно-щелочной баланс. В организме он поддерживается благодаря работе почек, буферных систем и лёгких. Если в крови понижается концентрация натрия, запускается РААС. Под влиянием альдостерона ионы возвращаются в кровь и соединяются с анионами, тем самым создают щелочную среду. Из организма выводятся кислоты в виде аммонийных солей (мочевины). Этот процесс способствует сохранению в организме необходимых минералов (натрия, калия, магния) и выведению токсинов.
Как только под влиянием РААС в крови из-за увеличения солей повышается осмотическое давление, стимулируется выработка вазопрессина, оказывается влияние на синтез альдостерона.
- При понижении концентрации хлорида натрия под воздействием гормонов удерживается в организме натрий и выводится вода. Так в организме сохраняется необходимое количество солей.
- Как только концентрация хлорида натрия повысилась, перестаёт работать РААС. В почечных клубочках происходит выведение избытка солей из организма.
Таким образом регулируется водно-солевой обмен и тем самым поддерживается:
- необходимый объём крови;
- нормальная концентрация натрия.
Кроме вазопрессина и альдостерона регуляцию водно-солевого баланса осуществляет и ангиотензин. Когда количество воды в крови снижается, он сужает стенки сосудов, чтобы временно поддержать нормальное артериальное давление (если объём крови недостаточный, возникает гипотензия) и обеспечить все органы необходимыми веществами. Также он влияет на центр жажды, расположенный в 3 желудочке головного мозга, из-за чего начинает хотеться пить. Как только в организм поступает необходимая жидкость и соли, перестаёт вырабатываться ренин. На этом работа РААС временно прекращается.
Если в организме произошёл сбой ренин-ангиотензин-альдостероновой системы, например, из-за:
- стеноза почечной артерии;
- и др. патологий.
Это приведёт к тому, что будет постоянно повышенное давление.
Кроме того, ангиотензин ІІ оказывает прямое воздействие на центральную нервную систему, возникает импульс, который буквально даёт команду гладкой мускулатуре сократиться. Сжимаются стенки сосудов, учащается сердцебиение, поднимается артериальное давление.
Изучение механизма действия РААС привело к тому, что были изобретены эффективные :
- блокаторы рецепторов к ангиотензину;
- ингибиторы АПФ.
Все эти медикаменты влияют на отдельные элементы цепочки выработки ренина, превращения ангиотензина, синтеза альдостерона. Естественно, они негативно влияют на работу системы и способствуют понижению артериального давления.
Вывод
 Механизм действия РААС
Механизм действия РААС РААС принимает активное участие в водно-солевом обмене, поддержании нормального давления и кислотно-щелочного баланса в крови. За считанные доли секунд вырабатывается ренин, ангиотензин и альдостерон, которые регулируют постоянный объём крови и необходимую концентрацию воды и солей. Однако и эта система может давать сбои, возникающие из-за болезней почек, надпочечников, а это приводит к патологическому повышению давления. Вот поэтому при гипертензии обязательно необходима консультация уролога, нефролога.
Общий видеообзор ренин-ангиотензин-альдостероновой системы: